


Tensofar®
REGISTRO SANITARIO:
INVIMA 2014M-0015279.
PRINCIPIO ACTIVO:
Losartán potásico
UNIDAD DE VENTA:
Tableta
Tabletas
(Losartán potásico)
COMPOSICIÓN: Cada tableta de TENSOFAR® 50 contiene 50 mg de losartán potásico, excipientes c.s. Cada tableta de TENSOFAR® 100 contiene 100 mg de losartán potásico, excipientes c.s.
INDICACIONES: Antihipertensivo (50 mg), Tratamiento de la hipertensión (100 mg)
VÍA DE ADMINISTRACIÓN: Oral
FORMA FARMACÉUTICA: Tableta
DOSIFICACIÓN: La dosis aconsejada es de 50 mg en una toma única diaria. En sujetos añosos (> 75 años) o con deterioro de la función hepática o renal se aconseja iniciar el tratamiento con 25 mg. En sujetos hipertensos refractarios o no respondedores a la dosis habitual se puede llegar a 100 mg diarios, pero no se lograron mejorías con dosis mayores.
CONTRAINDICACIONES (Tensofar 50 – 100 mg): Losartán está contraindicado en los pacientes que son hipersensibles a cualquier componente de este producto. La combinación de aliskiren con IECA o ARA II en pacientes con insuficiencia renal moderada-grave o diabetes está contraindicada, debido a un riesgo aumentado de hipotensión, sincope, hipercalcemia y cambios en la función renal (incluyendo fallo renal agudo) en comparación con la monoterapia. Contraindicado en el embarazo, Lactancia y niños menores de 15 años. Puede presentar ligero vértigo pasajero. ADVERTENCIAS: Toxicidad fetal. Embarazo: Categoría D. El uso de fármacos que actúan sobre el sistema renina angiotensina durante el 2º y 3er trimestre del embarazo reduce la función renal y aumenta la morbilidad y mortalidad fetal y neonatal. El oligohidramnios resultante puede estar asociado a hipoplasia pulmonar fetal y deformaciones esqueléticas. Los posibles efectos adversos neonatales incluyen hipoplasia craneana, anuria, hipotensión, insuficiencia renal, y muerte. Cuando se detecta el embarazo, discontinúe la administración de losartán tan pronto como sea posible. Estos efectos adversos se encuentran usualmente asociados con el uso de estos fármacos durante el 2º y 3er trimestre del embarazo. La mayoría de los estudios epidemiológicos que examinaron anormalidades fetales después de la exposición al uso de antihipertensivos en el primer trimestre no han distinguido entre fármacos que afectan al sistema renina angiotensina de otros agentes antihipertensivos. El manejo apropiado de la hipertensión materna durante el embarazo es importante para optimizar los desenlaces tanto para la madre como para el feto. En el caso no usual en que no haya una alternativa apropiada a la terapia con fármacos que afectan al sistema renina angiotensina para una paciente en particular, informe la madre de los riesgos potenciales para el feto. Lleve a cabo exámenes con ultrasonido seriados para evaluar el ambiente intra-amniótico. Si se observa oligohidramnios, discontinúe la administración de losartán, a menos que se considere necesario para salvar la vida de la madre. Las pruebas fetales pueden ser apropiadas, con base en las semanas de embarazo. Tanto pacientes como médicos deben estar al tanto, sin embargo, que el oligohidramnios pudiera no aparecer sino hasta después de que el feto ha sufrido lesiones irreversibles. Observe estrechamente a los infantes con historial de exposición in útero a losartán en busca de hipotensión, oliguria, e hiperkalemia. Se ha demostrado que el losartán potásico produce efectos adversos en fetos y recién nacidos de la rata, incluso disminución del peso corporal, retraso del desarrollo físico y del comportamiento, mortalidad y toxicidad renal. Con la excepción del aumento del peso neonatal (que se afectó a dosis de sólo 10 mg/kg al día), las dosis relacionadas con estos efectos superaron los 25 mg/kg al día (aproximadamente el triple de la dosis máxima recomendada para seres humanos de 100 mg, sobre la base de mg/m2). Estas observaciones se atribuyen a la exposición al fármaco al final del embarazo y durante la lactancia. En la leche de rata se demostró que había concentraciones significativas de losartán y de su metabolito activo en el plasma fetal de la rata durante la fase tardía del embarazo. Hipotensión: Pacientes con reducción del volumen En los pacientes con una marcada reducción del volumen intravascular (p. ej, aquellos tratados con diuréticos), puede producirse hipotensión sintomática tras el inicio del tratamiento con losartán. Esta situación debe corregirse antes de la administración de losartán o deberá emplearse una dosis inicial más baja. No utilizar terapia combinada con medicamentos que actúan sobre el SRA (IECA, ARA II o aliskiren), excepto en aquellos casos que se considere imprescindible. En estos casos, el tratamiento debe llevarse a cabo bajo la supervisión de un médico con experiencia en el tratamiento de este tipo de pacientes, vigilando estrechamente la función renal, el balance hidroelectrolítico y la tensión arterial. PRECAUCIONES GENERALES: Hipersensibilidad: Angioedema. Alteración de la función hepática Basándose en los datos farmacocinéticos que demuestran un aumento significativo de las concentraciones plasmáticas de losartán en los pacientes cirróticos, deberá administrarse la dosis más baja a los pacientes con deterioro de la función hepática. Insuficiencia renal Como consecuencia de la inhibición del sistema renina angiotensina aldosterona, se han comunicado cambios en la función renal en personas susceptibles tratadas con losartán; en algunos pacientes, estos cambios en la función renal fueron reversibles al interrumpir la terapia. En los pacientes cuya función renal pueda depender de la actividad del sistema renina angiotensina aldosterona (p. ej, pacientes con insuficiencia cardiaca congestiva grave), el tratamiento con inhibidores de la enzima convertidora de la angiotensina se ha asociado a oliguria y/o azotemia progresiva y (raramente) con insuficiencia renal aguda y/o muerte. Se han comunicado problemas similares con losartán. En los estudios con IECA en pacientes con estenosis unilateral o bilateral de la arteria renal, se han comunicado aumentos de la creatinina sérica o del nitrógeno ureico sanguíneo (NUS). Se han comunicado efectos similares con losartán; en algunos pacientes, estos efectos fueron reversibles al interrumpir la terapia. No se recomienda el uso de la terapia combinada de IECA con ARA II, en particular en pacientes con nefropatía diabética. Desequilibrio electrolítico Los desequilibrios electrolíticos son frecuentes en los pacientes con insuficiencia renal, con o sin diabetes, y deberán abordarse. En un estudio clínico realizado con pacientes diabéticos tipo 2 con proteinuria, la incidencia de hiperkalemia fue más alta en el grupo tratado con losartán, en comparación con el grupo que recibió placebo; sin embargo, pocos pacientes interrumpieron el tratamiento a causa de hiperkalemia. Información para los pacientes. Embarazo: Debe informarse a las pacientes del género femenino en edad de concebir acerca de las consecuencias de la exposición a losartán durante el embarazo. Discuta opciones de tratamiento con mujeres que planeen quedar embarazadas. Se debe indicar a estas pacientes que, en caso de embarazo, lo comuniquen a sus médicos lo antes posible. Suplementos de potasio: Debe indicarse a los pacientes tratados con losartán que no usen suplementos de potasio ni sustitutos de la sal que contenga potasio, sin consultar con el médico que les ha prescrito el tratamiento.
INTERACCIONES: El empleo de diuréticos ahorradores de potasio puede incrementar la potasemia. No se han registrado interacciones significativas con el empleo simultáneo de antagonistas del calcio, betabloqueantes y diuréticos tiazídicos. La combinación con hidroclorotiazida puede potenciar la respuesta hipotensora. La hemodiálisis no sirve para eliminar el losartán y su metabolito activo.
EFECTOS SECUNDARIOS: Por lo general presenta una óptima tolerancia, pero en algunos pacientes se han presentado ocasionalmente (< 1%) mareos, exantema cutáneo, hipotensión ortostática, valores elevados de TGP que se normalizaron al suspender el tratamiento. A diferencia de los inhibidores de la enzima convertidora de la angiotensina, la incidencia de tos seca es menor (3% vs. 10%) y equiparable al placebo. En general, el losartán es bien tolerado. Se han reportado algunos casos de reacciones anafilácticas y de angioedema en pacientes tratados con losartán, aunque desde el punto de vista teórico, los antagonistas del receptor AT1 no causan la acumulación de quininas. Los pocos pacientes que experimentaron angioedema con el losartán habían experimentado previamente esta reacción adversa con inhibidores de la ECA o con otros fármacos (incluyendo alergias a la penicilina y al ácido acetilsalicílico). En un estudio clínico realizado en los efectos adversos digestivos (diarrea 2.4% y dispepsia 1.3%) fueron ligeramente superiores a los del placebo. En menos de 1% de los pacientes, se observó hipotensión ortostática y síncope, y algunos efectos musculoesqueléticos detectados con una frecuencia algo mayor que el placebo fueron mialgia (1.1 vs 0.38% para el losartán y el placebo, respectivamente), calambres musculares (1 vs 0.9%) dolor de espalda (1.8 vs 1.2%) y dolor de piernas (1 vs 0%). Los efectos adversos sobre el sistema nervioso central son mareos (3.5%) e insomnio (1.4%). También se han comunicado cefalea, astenia y fatiga, pero éstos también fueron observados con mayor frecuencia en los pacientes tratados con placebo. Los efectos sobre el sistema respiratorio que se observaron con mayor frecuencia en los pacientes tratados con losartán, fueron congestión nasal (3.4 vs 3.3%), tos (3.4 vs 3.3%) e infecciones del tracto respiratorio superior (7.9 vs 6.9%). El losartán produce menos tos que los inhibidores de la enzima de conversión al no inhibir la quinasa II de la enzima convertidora que se cree es la responsable de la tos que estos fármacos producen en muchos pacientes. Otros efectos adversos que aparecieron con una frecuencia menor o igual que el placebo fueron las faringitis. En los estudios clínicos preliminares se observó azoemia en < 0.1% de los pacientes hipertensos tratados con el losartán. Sin embargo, en el estudio ELITE se produjo una disfunción renal en 10.5% de los pacientes con insuficiencia cardiaca que fueron tratados con losartán, igual que el captopril que también ocasionó disfunción renal en el 10.5% de los casos. Estos resultados sugieren que en los pacientes cuya función renal es dependiente en algún grado del sistema renina-angiotensina, la supresión de la angiotensina puede ocasionar una disfunción renal.
PROPIEDADES FARMACODINÁMICAS: Es el primer derivado de una nueva generación de fármacos llamados antagonistas de la angiotensina II (ATII) que desarrolla un gradual y prolongado efecto sobre los valores sistodiastólicos de sujetos hipertensos. Se trata de una sustancia sintética de estructura química original bifeniltetrazol, de naturaleza no peptídica, que por su semejanza estructural compite con el receptor específico de la angiotensina II inhibiendo de esta manera su unión con este agonista endógeno. Su elevada afinidad y especificidad in vitro e in vivo sobre los receptores AT 1 de la angiotensina II, localizados preferentemente en el músculo liso vascular y otras estructuras (miocardio, riñón, cerebro, suprarrenal) y su comportamiento como un antagonista puro sin efecto agonista parcial, lo destacan como un agente antihipertensivo eficaz y seguro.
CONDICIÓN DE VENTA: Venta con fórmula facultativa
PRESENTACIÓN: TENSOFAR® 50 mg caja por 30 tabletas. Reg. San. INVIMA 2014M-0015279. TENSOFAR® 100 mg. Caja por 30 tabletas. Reg. San. INVIMA 2014M-0015211.


Tensofar® H
REGISTRO SANITARIO:
INVIMA 2024M-0021405
INVIMA 2024M-0021398
INVIMA 2024M-0021397
PRINCIPIO ACTIVO:
contiene 100 mg de losartán potásico 12,5 mg de hidroclorotiazia, excipientes c.s
UNIDAD DE VENTA:
Tableta
Tensofar®H 100/25 mg
Tensofar® H 100/12,5 mg
Tensofar®H 50/12,5
Tabletas
(Losartán potásico, hidroclorotiazida)
COMPOSICIÓN: Cada tableta de TENSOFAR® H 50 100/25 contiene 100 mg de losartán potásico, 25 mg de hidroclorotiazida, excipientes c.s. Cada tableta de TENSOFAR® H 100/12,5 contiene 100 mg de losartán potásico 12,5 mg de hidroclorotiazia, excipientes c.s. Cada tableta de TENSOFAR® H 50/12,5 contiene 50 mg de losartán potásico, 12,5 mg de hidroclorotiazida, excipientes c.s.
INDICACIONES: Hipertensión:tratamiento de la hipertensión cuando el tratamiento inicial con losartán o hidroclorotiazida sola no resulta en un adecuado control de la presión arterial. En pacientes con hipertensión e hipertrofia ventricular izquierda, losartán, usualmente en combinación con hidroclorotiazida, y como consecuencia del adecuado control de la presión arterial reduce el riesgo de morbilidad y mortalidad cardiovascular medida por la incidencia combinada de muerte cardiovascular, apoplejía e infarto de miocardio en pacientes hipertensos con hipertrofia ventricular izquierda.
VÍA DE ADMINISTRACIÓN: Oral
FORMA FARMACÉUTICA: Tableta
DOSIFICACIÓN: Hipertensión: Losartán e hidroclorotiazida no está indicado como tratamiento inicial, excepto en pacientes cuya presión arterial no está controlada adecuadamente con losartán potásico o hidroclorotiazida solos. Se recomienda el ajuste de dosis con los componentes individuales (losartán e hidroclorotiazida).Cuando sea clínicamente apropiado, puede considerarse el cambio directo de la monoterapia a la combinación fija en pacientes cuya presión arterial no esté controlada adecuadamente. La dosis recomendada de mantenimiento de losartán/hidroclorotiazida es un comprimido de 50 mg/12,5 mg (50 mg de losartán/12,5 mg de HCTZ) una vez al día. Para los pacientes que no respondan adecuadamente a 50 mg de losartán /12,5 mg de HCTZ, la dosis puede aumentarse a un comprimido de losartán/hidroclorotiazida 100 mg/12,5 mg o 100 mg de losartán/25 mg de HCTZ una vez al día, según la respuesta clínica. La dosis máxima es un comprimido de losartán/hidroclorotiazida 100 mg/25 mg una vez al día. En general, el efecto antihipertensivo se alcanza a las tres o cuatro semanas de iniciar el tratamiento. Uso en pacientes con insuficiencia renal y en pacientes sometidos a hemodiálisis: No se requiere un ajuste en la dosis inicial en pacientes con insuficiencia renal moderada (p. ej. aclaramiento de creatinina 30-50 ml/min). No se recomiendan los comprimidos de losartán e hidroclorotiazida en pacientes sometidos a hemodiálisis. No deben usarse los comprimidos de losartán/HCTZ en pacientes con insuficiencia renal grave (p. ej. aclaramiento de creatinina < 30 ml/min). Uso en pacientes con depleción del volumen intravascular: Debe corregirse la depleción de volumen y/o sodio antes de la administración de los comprimidos de losartán/HCTZ. Uso en pacientes con insuficiencia hepática: losartán/HCTZ está contraindicado en pacientes con insuficiencia hepática grave. Uso en pacientes de edad avanzada: Normalmente no es necesario el ajuste de dosis en pacientes de edad avanzada. Uso en niños y adolescentes (< 18 años): No se ha establecido todavía la seguridad y eficacia de losartán/hidroclorotiazida en niños y adolescentes. Por tanto, no se debe administrar losartán/hidroclorotiazida a niños y adolescentes.
CONTRAINDICACIONES (Tensofar-H 100/25 y Tensofar-H 100/12,5): el producto está contraindicado en: pacientes con hipersensibilidad a cualquier componente de este producto. Pacientes con anuria. – pacientes con hipersensibilidad a otros medicamentos sulfonamídicos. El producto no debe ser administrado con aliskiren en pacientes con diabetes. PRECAUCIONES Y ADVERTENCIAS: losartán – hidroclorotiazida toxicidad fetal el uso de medicamentos que actúan sobre el sistema renina-angiotensina durante el segundo y tercer trimestre de embarazo, reduce la función renal fetal e incrementa la morbilidad y muerte fetal y neonatal. Oligohidramnios resultantes pueden ser asociados con hipoplasia pulmonar fetal y deformaciones esqueléticas. Los eventos adversos neonatales potenciales incluyen hipoplasia craneal, anuria, hipotensión, falla renal y muerte. Si la paciente queda en estado de embarazo, se debe suspender la administración del producto lo más pronto posible. Hipersensibilidad: angioedema. Insuficiencia hepática o renal no se recomienda administrar el producto en pacientes con insuficiencia hepática o con insuficiencia renal severa (depuración de creatinina 30 ml/min). Losartán deterioro de la función renal como consecuencia de la inhibición del sistema renina-angiotensina en sujetos susceptibles, se han reportado cambios en la función renal incluyendo insuficiencia renal; estos cambios en la función renal se pueden revertir al suspender la terapia. Otros medicamentos que afectan el sistema renina-angiotensina pueden aumentar la urea sanguínea y la creatinina sérica en pacientes con estenosis bilateral de las arterias renales o de la arteria de un riñón único. Efectos similares se han reportado con losartán; estos cambios en la función renal se pueden revertir al descontinuar la terapia. No se recomienda el uso de la terapia combinada de IECA con ARA II, en particular en pacientes con nefropatía diabética. Incremento en el potasio sérico el uso concomitante de otros medicamentos que pueden aumentar el potasio sérico puede provocar hipercalemia. Hidroclorotiazida hipotensión y desequilibrio hidroelectrolítico como ocurre con todos los tratamientos antihipertensivos, algunos pacientes pueden presentar hipotensión sintomática. Se debe vigilar la aparición de signos clínicos de desequilibrio hídrico o electrolítico, como por ejemplo disminución de volumen, hiponatremia, alcalosis hipoclorémica, hipomagnesemia o hipopotasemia, que pueden ocurrir si hay diarrea o vómito recurrentes. En esos pacientes se deben efectuar determinaciones periódicas de los electrólitos séricos en intervalos adecuados. Efectos metabólicos y endocrinológicos las tiazidas pueden disminuir la tolerancia a la glucosa. Puede ser necesario ajustar la dosis de los agentes antidiabéticos, incluyendo la insulina. Las tiazidas pueden disminuir la excreción urinaria de calcio y causar aumentos intermitentes y leves del calcio sérico. La hipercalcemia marcada puede ser indicio de un hiperparatiroidismo oculto. Se debe suspender las tiazidas antes de realizar pruebas de función paratiroidea. Los aumentos en los niveles de colesterol y triglicéridos pueden estar asociados con la terapia diurética con tiazida. La terapia con tiazidas puede precipitar hiperuricemia y/o gota en ciertos pacientes. Como losartán disminuye el ácido úrico, su combinación con hidroclorotiazida atenúa la hiperuricemia inducida por el diurético. Cáncer de piel no melanoma en estudios epidemiológicos se ha observado un mayor riesgo de cáncer de piel no melanoma (carcinoma de células basales [BCC, por sus siglas en inglés] y carcinoma de células escamosas [SCC, por sus siglas en inglés]) con un incremento de la dosis acumulativa de hidroclorotiazida. Las acciones foto-sensibilizantes de la hidroclorotiazida podrían actuar como un posible mecanismo para el cáncer de piel no melanoma. Los pacientes que toman hidroclorotiazida deben ser informados sobre el riesgo de cáncer de piel no melanoma y se les debe recomendar que tomen medidas preventivas para reducir la exposición al sol y a los rayos uva artificiales. Los pacientes deben revisar regularmente su piel en busca de nuevas lesiones e informar rápidamente a sus médicos de lesiones cutáneas sospechosas para su evaluación. El uso de hidroclorotiazida también puede ser reconsiderado en pacientes que hayan experimentado cáncer de piel no melanoma previo. Otras en pacientes que están recibiendo tiazidas pueden ocurrir reacciones de hipersensibilidad, con o sin antecedentes de alergia o de asma bronquial. Se ha reportado exacerbación o activación del lupus eritematoso sistémico durante el uso de tiazidas.
CONTRAINDICACIONES (Tensofar-H 50/12,5): está contraindicado en los pacientes con hipersensibilidad a cualquiera de los componentes de este producto. Pacientes con anuria, pacientes hipersensibles a otros medicamentos sulfonamidicos, embarazo, lactancia, menores de 18 años. Pacientes con daño hepático o renal. La combinación de aliskireno con IECA o ARA II en pacientes con insuficiencia renal moderada a grave o diabetes esta contraindicada. ADVERTENCIAS: toxicidad fetal embarazo categoría D. El uso de fármacos que actúan sobre el sistema renina-angiotensina durante el segundo y tercer trimestre del embarazo reduce la función renal y aumenta la morbilidad y mortalidad fetal y neonatal. El oligohidramnios resultante puede estar asociado a hipoplasia del pulmón fetal y a deformaciones esqueléticas fetales. Los posibles efectos adversos neonatales incluyen hipoplasia del cráneo, anuria, hipotensión, insuficiencia renal, y muerte. Cuando se detecta el embarazo, discontinúe el producto tan pronto como sea posible. Estos desenlaces adversos se encuentran usualmente asociados al uso de estos fármacos en el segundo y tercer trimestre del embarazo. La mayoría de los estudios epidemiológicos que han examinado anormalidades fetales después de la exposición al uso de antihipertensivos en el primer trimestre no han distinguido los fármacos que afectan al sistema renina-angiotensina de otros agentes antihipertensivos. El manejo apropiado de la hipertensión materna durante el embarazo es importante para optimizar los desenlaces tanto para la madre como para el feto. En el caso inusual de que no haya alternativas apropiadas a la terapia con fármacos que afectan el sistema renina-angiotensina para una paciente en particular, informe a la madre del riesgo potencial para el feto. Lleve a cabo exámenes seriados por ultrasonido con el fin de evaluar el ambiente intra-amniótico. Si se observa oligohidramnios, suspenda el tratamiento con el producto a menos que se considere de importancia vital para la madre. Puede ser apropiado hacer una evaluación fetal, con base en las semanas de embarazo. Las pacientes y los médicos deben saber, sin embargo, que el oligohidramnios puede no aparecer hasta que el feto tenga lesiones irreversibles. Vigile cercanamente a los infantes con historial de exposición in útero al producto en busca de hipotensión, oliguria, e hiperkalemia no se observaron datos de teratogenia en ratas ni conejos tratados con una dosis máxima de losartán potásico de 10 mg/kg/día en combinación con 2.5 mg/kg/día de hidroclorotiazida. A estas dosis, las exposiciones respectivas (AUC) al losartán, su metabolito activo y la hidroclorotiazida en los conejos fueron aproximadamente 5, 1.5 y 1.0 veces más altas que las alcanzadas en humanos con 100 mg de losartán combinados con 25 mg de hidroclorotiazida. Los valores del AUC para el losartán, su metabolito activo y la hidroclorotiazida, extrapolados a partir de los datos obtenidos con la administración de losartán a ratas en dosis de 50 mg/kg/día en combinación con 12.5 mg/kg/día de hidroclorotiazida, fueron aproximadamente 6, 2 y 2 veces más altos que los alcanzados en humanos con 100 mg de losartán combinados con 25 mg de hidroclorotiazida. Se observó toxicidad fetal en ratas, con un ligero aumento de las costillas supernumerarias, cuando las hembras recibieron tratamiento antes de la gestación y durante la misma con 10 mg/kg/día de losartán combinado con 2.5 mg/kg/día de hidroclorotiazida. Tal como se observó también en los estudios con losartán solo, se produjeron efectos adversos fetales y neonatales, incluidos reducción del peso corporal, toxicidad renal y muerte, cuando se trató a las ratas preñadas durante las etapas finales de la gestación y/o la lactancia con 50 mg/kg/día de losartán combinados con 12.5 mg/kg/día de hidroclorotiazida. Las AUC respectivas para el losartán, su metabolito activo y la hidroclorotiazida a estas dosis en las ratas fueron aproximadamente 35, 10 y 10 veces más altas que las alcanzadas en humanos con la administración de 100 mg de losartán combinados con 25 mg de hidroclorotiazida. No se observaron datos indicativos de daño fetal cuando se administró hidroclorotiazida sin losartán a ratones y ratas gestantes durante sus periodos respectivos de mayor organogénesis a dosis de hasta 3,000 y 1,000 mg/kg/día, respectivamente. Las tiazidas atraviesan la barrera placentaria y aparecen en la sangre del cordón. Existe riesgo de ictericia fetal o neonatal, trombocitopenia y posiblemente otras reacciones adversas observadas en adultos. No utilizar terapia combinada con medicamentos que actúan sobre el SRA ( IECA, ARA II o aliskireno), excepto en aquellos casos que se considere imprescindible. En estos casos, el tratamiento debe llevarse a cabo bajo la supervisión de un médico con experiencia en el tratamiento de este tipo de pacientes, vigilando estrechamente la función renal, el balance hidroelectrolítico y la tensión arterial. Hipotensión – pacientes con marcada reducción de volumen en los pacientes con una marcada reducción del volumen intravascular (p. ej., aquellos tratados con diuréticos), puede producirse hipotensión sintomática tras el inicio de la terapia con el producto. Esta situación debe corregirse antes de la administración del producto. Deterioro de la función hepática losartán potásico – hidroclorotiazida no se recomienda en pacientes con deterioro de la función hepática que precisen un ajuste progresivo de la dosis de losartán. La dosis de inicio más baja de losartán recomendada para los pacientes con deterioro de la función hepática no puede administrarse cuando se usa el producto. Hidroclorotiazida las tiazidas deben usarse con precaución en los pacientes con deterioro de la función hepática o hepatopatía progresiva, dado que alteraciones muy pequeñas del equilibrio hidroelectrolítico pueden precipitar un coma hepático. Reacciones de hipersensibilidad pueden almacenar a temperatura inferior a 30ºc en su envase y empaque original. Producirse reacciones de hipersensibilidad a la hidroclorotiazida en pacientes con o sin antecedentes de alergia o asma bronquial, pero son más probables en los pacientes con dichos antecedentes. Lupus eritematoso sistémico se ha comunicado que los diuréticos tiazídicos pueden causar una exacerbación o activación de un lupus eritematoso sistémico. Interacciones con el litio como norma general, el litio no debe administrarse con tiazidas. Vigile los niveles séricos de litio en pacientes que reciben litio e hidroclorotiazida. Miopía aguda y glaucoma secundario de ángulo cerrado la hidroclorotiazida, una sulfonamida, puede ocasionar una reacción idiosincrática, que resulta en miopía aguda transitoria y glaucoma agudo de ángulo cerrado. Los síntomas incluyen un inicio agudo de disminución de la agudeza visual o dolor ocular y típicamente se presentan dentro de horas a semanas del inicio del fármaco. El glaucoma agudo de ángulo cerrado no tratado puede llevar a una pérdida permanente de la visión. El tratamiento primario es la discontinuación de la hidroclorotiazida tan rápidamente como sea posible. Debe considerarse la necesidad de un pronto tratamiento médico o quirúrgico si la presión intraocular permanece sin control. Los factores de riesgo para desarrollar glaucoma agudo de ángulo cerrado pueden incluir un historial de alergia a las sulfonamidas o a penicilinas. PRECAUCIONES: hipersensibilidad: angioedema. Losartán potásico – hidroclorotiazida en los ensayos clínicos a doble ciego con varias dosis de losartán potásico e hidroclorotiazida, la incidencia de pacientes hipertensos que presentaron hipopotasemia (potasio sérico <3.5 meq/l) fue del 6.7% frente al 3.5% para el placebo; la incidencia de hiperpotasemia (potasio sérico >5.7 meq/l) fue del 0.4%. Ningún paciente interrumpió el tratamiento a causa de un aumento o un descenso en el potasio sérico. El descenso medio del potasio sérico en los pacientes tratados con varias dosis de losartán e hidroclorotiazida fue de 0.123 mEq/l. En los pacientes tratados con varias dosis de losartán e hidroclorotiazida, también se observó una reducción relacionada con la dosis de la respuesta hipopotasémica a la hidroclorotiazida a medida que se aumentaba la dosis de losartán, así como una reducción relacionada con la dosis del ácido úrico sérico con dosis crecientes de losartán. Hidroclorotiazida deben realizarse determinaciones periódicas de los electrolitos séricos en intervalos adecuados para detectar posibles desequilibrios electrolíticos. Todos los pacientes tratados con tiazidas se deben examinar en busca de signos clínicos de desequilibrio hídrico o electrolítico: hiponatremia, alcalosis hipoclorémica e hipopotasemia. Las determinaciones de electrolitos en suero y en orina son especialmente importantes si el paciente vomita excesivamente o está recibiendo líquidos por vía parenteral. Los signos y síntomas de alerta de desequilibrio hidroelectrolítico, independientemente de su causa, son sequedad de boca, sed, debilidad, letargo, somnolencia, inquietud, confusión, crisis epilépticas, dolores o calambres musculares, fatiga muscular, hipotensión, oliguria, taquicardia y alteraciones gastrointestinales como náuseas y vómitos. Puede producirse hipopotasemia, especialmente cuando existe una diuresis enérgica, en los casos de cirrosis grave o tras una terapia prolongada. La interferencia con una adecuada ingesta oral de electrolitos también contribuye a la hipopotasemia. La hipopotasemia puede causar arritmias cardiacas y también sensibilizar al corazón o exagerar su respuesta a los efectos tóxicos de la digital (p.ej., aumento de la irritabilidad ventricular). Aunque todos los déficits de cloro son generalmente de grado leve y no suelen requerir tratamiento específico excepto en circunstancias extraordinarias (como, p.ej., enfermedad hepática o renal), puede ser necesario reponer dicho electrolito para tratar una alcalosis metabólica. En clima calido, puede producirse hiponatremia dilucional en los pacientes con edema; el tratamiento pertinente es la restricción de líquidos y no la administración de sal, excepto en los raros casos en los que la hiponatremia sea potencialmente mortal. Si existe una reducción real marcada de sal, el tratamiento de elección es el reemplazo de la misma tal como resulte necesario. Puede producirse hiperuricemia o precipitarse una gota franca en algunos pacientes tratados con tiazidas. Dado que el losartán reduce el ácido úrico, su combinación con hidroclorotiazida atenúa la hiperuricemia inducida por dicho diurético. En los pacientes diabéticos, puede ser necesario ajustar la dosis de insulina o de antidiabéticos orales. Los diuréticos tiazídicos pueden causar hiperglucemia, por lo que una diabetes mellitus latente puede hacerse franca durante un tratamiento con estos fármacos. Los efectos antihipertensivos del medicamento pueden potenciarse en los pacientes sometidos a una simpatectomía. En caso de constatarse un deterioro progresivo de la función renal, es necesario considerar la interrupción o la suspensión de la terapia con diuréticos. Las tiazidas han mostrado aumentar la excreción urinaria de magnesio, lo que puede dar lugar a una hipomagnesemia. Las tiazidas pueden reducir la excreción urinaria de calcio. Asimismo, pueden causar una elevación ligera e intermitente del calcio sérico en ausencia de trastornos conocidos del metabolismo del calcio. Una marcada hipercalcemia puede ser un dato indicativo de un hiperparatiroidismo oculto. Las tiazidas deben interrumpirse antes de la realización de pruebas de función paratiroidea. La terapia con diuréticos tiazídicos puede asociarse con un aumento de las concentraciones de colesterol y triglicéridos. Deterioro de la función renal como consecuencia de la inhibición del sistema renina-angiotensina-aldosterona, se han comunicado cambios en la función renal en personas susceptibles tratadas con losartán; en algunos pacientes, estos cambios en la función renal fueron reversibles al interrumpir la terapia. En los pacientes cuya función renal pueda depender de la actividad del sistema reninaangiotensinaaldosterona (p.ej., pacientes con insuficiencia cardiaca congestiva grave), el tratamiento con inhibidores de la enzima convertidora de la angiotensina se ha asociado a oliguria y/o azotemia progresiva y (raramente) con insuficiencia renal aguda y/o muerte. Se han comunicado problemas similares con el losartán. En los estudios con IECA en pacientes con estenosis unilateral o bilateral de la arteria renal, se han comunicado aumentos de la creatinina sérica o del nitrógeno ureico sanguíneo (nus). Se han comunicado efectos similares con el losartán; en algunos pacientes, estos efectos fueron reversibles al interrumpir la terapia. Las tiazidas deben usarse con precaución en los pacientes con nefropatías graves. En los pacientes con enfermedad renal, las tiazidas pueden precipitar una azotemia. En los pacientes con deterioro de la función renal pueden aparecer efectos por acumulación del fármaco. No se recomienda el uso de la terapia combinada de IECA con ARA II, en particular en pacientes con nefropatía diabética. Información para los pacientes embarazo: debe informarse a las pacientes en edad reproductiva sobre las consecuencias de la exposición a losartan + hidroclorotiazida durante el embarazo. Discuta opciones de tratamiento con mujeres que planean quedar embarazadas. Se debe indicar a estas pacientes que, en caso de embarazo, lo comuniquen a sus médicos lo antes posible. Hipotensión sintomática: debe advertirse a los pacientes tratados con losartan + hidroclorotiazida que pueden sufrir sensación de desvanecimiento, especialmente durante los primeros días de terapia, que deben comunicar al médico que les ha prescrito el tratamiento. Debe indicarse a los pacientes que, en caso de síncope, es necesario interrumpir el tratamiento con el producto hasta consultar al médico. Debe advertirse a todos los pacientes que la ingesta inadecuada de líquidos, la sudoración excesiva, la diarrea o los vómitos pueden llevar a un descenso excesivo de la presión arterial, con las mismas consecuencias de sensación de desvanecimiento y posible síncope. Suplementos de potasio: debe indicarse a los pacientes tratados con el producto que no usen suplementos de potasio ni sustitutos de la sal que contenga potasio sin consultar con el médico que les ha prescrito el tratamiento. Cáncer de piel no melanoma en estudios epidemiológicos se ha observado un mayor riesgo de cáncer de piel no melanoma (carcinoma de células basales [BCC, por sus siglas en inglés] y carcinoma de células escamosas [SCC, por sus siglas en inglés]) con un incremento de la dosis acumulativa de hidroclorotiazida. Las acciones fotosensibilizantes de la hidroclorotiazida podrían actuar como un posible mecanismo para el cáncer de piel no melanoma. Los pacientes que toman hidroclorotiazida deben ser informados sobre el riesgo de cáncer de piel no melanoma y se les debe recomendar que tomen medidas preventivas para reducir la exposición al sol y a los rayos uva artificiales. Los pacientes deben revisar regularmente su piel en busca de nuevas lesiones e informar rápidamente a sus médicos de lesiones cutáneas sospechosas para su evaluación. El uso de hidroclorotiazida también puede ser reconsiderado en pacientes que hayan experimentado cáncer de piel no melanoma previo.
INTERACCIONES: Losartán: Se ha notificado que rifampicina y fluconazol reducen los metabolitos del principio activo. No se han evaluado las consecuencias clínicas de estas interacciones. Al igual que ocurre con otros medicamentos que bloquean la angiotensina II o sus efectos, el uso concomitante de otros diuréticos ahorradores de potasio (p. ej., espironolactona, triamtereno, amilorida), suplementos de potasio, sustitutos de la sal que contienen potasio, u otros medicamentos que puedan aumentar el potasio sérico (p. ej. medicamentos que contengan trimetoprim), puede provocar aumentos de los niveles plasmáticos de potasio. Por tanto, la administración conjunta con estos medicamentos no es aconsejable. Litio: al igual que ocurre con otros medicamentos que afectan a la excreción de sodio, la excreción de litio puede reducirse. Por tanto, deben vigilarse estrechamente las concentraciones séricas de litio si se coadministran las sales de litio con antagonistas de los receptores de la angiotensina II. Cuando los antagonistas de la angiotensina II se administran junto con AINEs (p. ej. inhibidores selectivos de la COX-2, ASA a dosis antiinflamatorias y AINEs no selectivos), puede producirse una reducción del efecto antihipertensivo. El uso concomitante de antagonistas de la angiotensina II o diuréticos y AINEs puede conducir a una elevación del riesgo de empeoramiento de la función renal, incluyendo posible fallo renal agudo y un aumento de los niveles plasmáticos de potasio, especialmente en pacientes con función renal reducida preexistente. Esta combinación debe administrarse con precaución, especialmente en pacientes de edad avanzada. Los pacientes deben ser adecuadamente hidratados y se debe vigilar su función renal tras el inicio del tratamiento concomitante y posteriormente de forma periódica. En algunos pacientes con la función renal comprometida que están siendo tratados con medicamentos antiinflamatorios no esteroideos, incluyendo los inhibidores selectivos de la COX-2, la coadministración de antagonistas de los receptores de la angiotensina II puede llevar a un empeoramiento de la función renal. Estos cambios normalmente son reversibles. Los datos de los estudios clínicos han demostrado que el bloqueo dual del sistema renina-angiotensina-aldosterona (SRAA) mediante el uso combinado de inhibidores de la enzima convertidora de angiotensina, antagonistas de los receptores de angiotensina II o aliskirén, se asocia con una mayor frecuencia de acontecimientos adversos tales como hipotensión, hiperpotasemia y disminución de la función renal (incluyendo insuficiencia renal aguda) en comparación con el uso de un solo agente con efecto sobre el SRAA. Otros medicamentos que inducen hipotensión como antidepresivos tricíclicos, antipsicóticos, baclofeno, amifostina: el uso concomitante de losartán con estos medicamentos que disminuyen la presión arterial, como efecto principal o adverso, pueden aumentar el riesgo de hipotensión. Hidroclorotiazida Cuando se administran simultáneamente, los siguientes medicamentos pueden interactuar con los diuréticos tiazídicos: Alcohol, barbitúricos, narcóticos o antidepresivos: puede producirse potenciación de la hipotensión ortostática. Antidiabéticos (medicamentos orales e insulina): el tratamiento con tiazidas puede afectar a la tolerancia de la glucosa. Puede ser necesario el ajuste de la dosis del medicamento antidiabético. La metformina debe usarse con precaución debido al riesgo de acidosis láctica inducida por posible fallo renal funcional ligado a hidroclorotiazida. Otros antihipertensivos: efecto aditivo. Resinas colestiramina y colestipol: en presencia de resinas de intercambio aniónico se altera la absorción de hidroclorotiazida. Las dosis únicas de las resinas colestiramina o colestipol se unen a la hidroclorotiazida y reducen su absorción en el tubo digestivo hasta en un 85 y 43 %, respectivamente. Corticosteroides, ACTH: depleción de electrolitos intensificada, en particular la hipopotasemia. Aminas presoras (p. ej., adrenalina): posible disminución de la respuesta a las aminas presoras, pero no suficiente para excluir su empleo. Relajantes no despolarizantes del músculo esquelético (p. ej., tubocurarina): posible aumento de la respuesta al relajante muscular. Litio: los medicamentos diuréticos reducen el aclaramiento renal de litio y añaden un elevado riesgo de toxicidad por litio; no se recomienda su uso concomitante. Medicamentos en tratamiento de la gota (probenecid, sulfinpirazona y alopurinol): pueden ser necesarios ajustes de dosis de medicamentos uricosúricos ya que hidroclorotiazida puede aumentar el nivel sérico de ácido úrico. Pueden ser necesarios aumentos de la dosis de probenecid o de sulfinpirazona. La coadministración de una tiazida puede aumentar la incidencia de reacciones de hipersensibilidad al alopurinol. Anticolinérgicos (p. ej. atropina, biperideno): aumento de la biodisponibilidad de los diuréticos tipo tiazida al disminuir la motilidad gastrointestinal y la velocidad de vaciado gástrico. Citotóxicos (p. ej. ciclofosfamida, metotrexato): las tiazidas pueden reducir la excreción renal de medicamentos citotóxicos y potenciar su efecto mielosupresor. Salicilatos: en caso de dosis altas de salicilatos, hidroclorotiazida puede potenciar el efecto tóxico de los salicilatos sobre el sistema nervioso central. Metildopa: ha habido informes aislados de anemia hemolítica produciéndose con el uso concomitante de hidroclorotiazida y metildopa. Ciclosporina: el tratamiento concomitante con ciclosporina puede aumentar el riesgo de hiperuricemia y complicaciones tipo gota. Glucósidos digitálicos: la hipopotasemia e hipomagnesemia inducida por las tiazidas puede favorecer la aparición de arritmias cardiacas inducidas por digitálicos. Medicamentos afectados por las alteraciones del potasio sérico: Se recomienda la vigilancia periódica del potasio sérico y electrocardiograma (ECG) cuando losartán/ hidroclorotiazida se administra con medicamentos afectados por alteraciones del potasio sérico (p. ej. glucósidos digitálicos y antiarrítmicos) y con los siguientes medicamentos que inducen torsades de pointes (taquicardia ventricular) (incluyendo algunos antiarrítmicos), al ser la hipopotasemia un factor que predispone a torsades de pointes: •Antiarrítmicos clase Ia (p. ej. quinidina, hidroquinidina, disopiramida). •Antiarrítmicos clase III (p. ej. amiodarona, sotalol, dofetilida, ibutilida). •Antipsicóticos (p. ej. tioridazina, clorpromazina, levomepromazina, trifluoperazina, ciamemazina, sulpirida, sultoprida, amisulprida, tiaprida, pimozida, haloperidol, droperidol). •Otros (p. ej. bepridil, cisaprida, difemanilo, eritromicina IV, halofantrina, mizolastina, pentamidina, terfenadina, vincamina IV). Sales de calcio: los diuréticos tiazídicos pueden aumentar las concentraciones séricas de calcio debido a una excreción reducida. Si se prescriben suplementos de calcio, se deben vigilar las concentraciones séricas de calcio y en consecuencia, ajustar la dosis de calcio. Interacciones con pruebas de laboratorio: debido a sus efectos sobre el metabolismo del calcio, las tiazidas pueden interferir con las pruebas de función paratiroidea. Carbamazepina: riesgo de hiponatremia sintomática. Se requiere vigilancia clínica y biológica. Medios de contraste yodados: en caso de deshidratación inducida por diuréticos, hay un mayor riesgo de insuficiencia renal aguda, especialmente con dosis altas de medicamentos con yodo. Se debe rehidratar a los pacientes antes de la administración. Amfotericina B (parenteral), corticoesteroides, ACTH, laxantes estimulantes o glicirricina (que se encuentra en el regaliz) La hidroclorotiazida puede intensificar el desequilibrio electrolítico, especialmente la hipopotasemia.
EFECTOS SECUNDARIOS: Por lo general presenta una óptima tolerancia, pero en algunos pacientes se han presentado ocasionalmente (< 1%) mareos, exantema cutáneo, hipotensión ortostática, valores elevados de TGP que se normalizaron al suspender el tratamiento. A diferencia de los inhibidores de la enzima convertidora de la angiotensina, la incidencia de tos seca es menor (3% vs. 10%) y equiparable al placebo. En general, el losartán es bien tolerado. Se han reportado algunos casos de reacciones anafilácticas y de angioedema en pacientes tratados con losartán, aunque desde el punto de vista teórico, los antagonistas del receptor AT1 no causan la acumulación de quininas. Los pocos pacientes que experimentaron angioedema con el losartán habían experimentado previamente esta reacción adversa con inhibidores de la ECA o con otros fármacos (incluyendo alergias a la penicilina y al ácido acetilsalicílico). En un estudio clínico realizado en los efectos adversos digestivos (diarrea 2.4% y dispepsia 1.3%) fueron ligeramente superiores a los del placebo. En menos de 1% de los pacientes, se observó hipotensión ortostática y síncope, y algunos efectos musculoesqueléticos detectados con una frecuencia algo mayor que el placebo fueron mialgia (1.1 vs 0.38% para el losartán y el placebo, respectivamente), calambres musculares (1 vs 0.9%) dolor de espalda (1.8 vs 1.2%) y dolor de piernas (1 vs 0%). Los efectos adversos sobre el sistema nervioso central son mareos (3.5%) e insomnio (1.4%). También se han comunicado cefalea, astenia y fatiga, pero éstos también fueron observados con mayor frecuencia en los pacientes tratados con placebo. Los efectos sobre el sistema respiratorio que se observaron con mayor frecuencia en los pacientes tratados con losartán, fueron congestión nasal (3.4 vs 3.3%), tos (3.4 vs 3.3%) e infecciones del tracto respiratorio superior (7.9 vs 6.9%). El losartán produce menos tos que los inhibidores de la enzima de conversión al no inhibir la quinasa II de la enzima convertidora que se cree es la responsable de la tos que estos fármacos producen en muchos pacientes. Otros efectos adversos que aparecieron con una frecuencia menor o igual que el placebo fueron las faringitis. En los estudios clínicos preliminares se observó azoemia en < 0.1% de los pacientes hipertensos tratados con el losartán. Sin embargo, en el estudio ELITE se produjo una disfunción renal en 10.5% de los pacientes con insuficiencia cardiaca que fueron tratados con losartán, igual que el captopril que también ocasionó disfunción renal en el 10.5% de los casos. Estos resultados sugieren que en los pacientes cuya función renal es dependiente en algún grado del sistema renina-angiotensina, la supresión de la angiotensina puede ocasionar una disfunción renal. Según los datos disponibles de los estudios epidemiológicos, se ha observado una asociación entre el cáncer de piel no melanoma (BCC y SCC) e hidroclorotiazida, dependiente de la dosis acumulada. El estudio más grande incluyó una población compuesta por 71,533 casos de BCC y 8,629 casos de SCC ligados con 1,430,833 y 172,462 controles de población, respectivamente. El alto uso acumulativo de hidroclorotiazida (≥ 50,000 mg) se asoció con una razón de probabilidades ajustada (odds ratio, OR) de 1.29 (IC 95%: 1.23-1.35) para BCC y 3.98 (IC 95%: 3.68-4.31) para SCC. Se observó una relación de dosis-respuesta acumulativa tanto para BCC como para SCC. Otro estudio evaluó la asociación entre el cáncer de labio (SCC) y la exposición a la hidroclorotiazida: 633 casos de cáncer de labio fueron ligados con 63,067 controles de población.
PROPIEDADES FARMACODINÁMICAS: Losartan: Es el primer derivado de una nueva generación de fármacos llamados antagonistas de la angiotensina II (ATII) que desarrolla un gradual y prolongado efecto sobre los valores sistodiastólicos de sujetos hipertensos. Se trata de una sustancia sintética de estructura química original bifeniltetrazol, de naturaleza no peptídica, que por su semejanza estructural compite con el receptor específico de la angiotensina II inhibiendo de esta manera su unión con este agonista endógeno. Su elevada afinidad y especificidad in vitro e in vivo sobre los receptores AT 1 de la angiotensina II, localizados preferentemente en el músculo liso vascular y otras estructuras (miocardio, riñón, cerebro, suprarrenal) y su comportamiento como un antagonista puro sin efecto agonista parcial, lo destacan como un agente antihipertensivo eficaz y seguro. Hidroclorotiazida: es una benzotiadiazina (tiazida) diurética. Las tiazidas diuréticas actúan principalmente en el túbulo contorneado distal renal inhibiendo la reabsorción de sodio y cloro. Se desconoce el mecanismo antihipertensivo de hidroclorotiazida. El mecanismo del efecto antihipertensivo de las tiazidas puede estar relacionado con la excreción y la redistribución del sodio del organismo.
CONDICIÓN DE VENTA: Venta con fórmula facultativa
PRESENTACIÓN: TENSOFAR® H 100/25 mg caja por 10 tabletas. Reg. San. INVIMA 2024M-0021405. TENSOFAR® H 100/12,5 mg caja por 10 tabletas. Reg. San. INVIMA 2024M-0021397. TENSOFAR® H 50/12,5 mg caja por 10 tabletas. Reg. San. INVIMA 2024M-0021398.

Acidrine® Tabletas
REGISTRO SANITARIO:
INVIMA 2023M-0020877
PRINCIPIO ACTIVO:
Atorvastatina
UNIDAD DE VENTA:
Tabletas
Inhibidores HMG CoA Reductasa
(Atorvastatina)
COMPOSICIÓN: Cada tableta recubierta contiene 40 mg de atorvastatina, excipientes c.s.
INDICACIONES: la atorvastatina está indicada como coadyuvante de la dieta en el manejo de las dislipoproteinemias. Útil en pacientes con múltiples factores de riesgo para enfermedad cardíaca coronaria, las cuales pueden incluir diabetes mellitus, historia de trombosis u otra enfermedad cerebrovascular o enfermedad cardíaca coronaria asintomática, para disminuir el riesgo de infarto de miocardio no fatal y trombosis no fatal. Atorvastatina también está indicada para la reducción del colesterol total y colesterol LDL en pacientes con hipercolesterolemia familiar homocigótica y heterocigótica. Reduce el riesgo de infarto del miocardio no fatal, eventos cerebrales fatales y no fatales, angina, procedimientos de revascularización y necesidad de hospitalización por insuficiencia cardíaca congestiva en adultos con enfermedad coronaria clínicamente evidente, con niveles de colesterol controlado. Está indicada en pacientes con enfermedad vascular periférica o enfermedad cardíaca coronaria asintomática para disminuir el riesgo de infarto al miocardio no fatal y trombosis no fatal. Uso pediátrico para niños mayores de 6 años.
VÍA DE ADMINISTRACIÓN: Oral
FORMA FARMACÉUTICA: tableta recubierta.
POSOLOGÍA Y DOSIFICACIÓN: La dosis inicial habitual es de 10 mg una vez al día.
El ajuste de la dosis se debe hacer a intervalos de 4 o más semanas. La dosis máxima es de 80 mg una vez al día. Hipercolesterolemia primaria e hiperlipidemia combinada (mixta) La mayoría de los pacientes se controlan con Atorvastatina 10 mg administrado una vez al día. La respuesta terapéutica se observa al cabo de 2 semanas y habitualmente se alcanza la máxima respuesta terapéutica a las 4 semanas. La respuesta se mantiene durante el tratamiento crónico. Hipercolesterolemia familiar heterocigótica Los pacientes deben iniciar el tratamiento con 10 mg de Atorvastatina al día. Las dosis deben individualizarse y ajustarse cada 4 semanas hasta los 40 mg al día. Posteriormente, la dosis puede aumentarse hasta un máximo de 80 mg al día o se puede combinar 40 mg de atorvastatina una vez al día con un secuestrante de ácidos biliares. Hipercolesterolemia familiar homocigótica: Sólo se dispone de datos limitados. La dosis de atorvastatina en pacientes con hipercolesterolemia familiar homocigótica es de 10 a 80 mg al día. Atorvastatina debe utilizarse en terapia combinada con otros tratamientos hipolipemiantes (por ejemplo, aféresis de las LDL) en estos pacientes o si no se dispone de estos tratamientos. Prevención de la enfermedad cardiovascular: en los estudios en prevención primaria la dosis fue 10 mg/día. Pueden ser necesarias dosis mayores a fin de alcanzar los niveles de colesterol LDL de acuerdo con las guías actuales. Insuficiencia renal No es necesario un ajuste de la dosis. Insuficiencia hepática: atorvastatina se debe utilizar con precaución en pacientes con insuficiencia hepática. Atorvastatina está contraindicado en pacientes con enfermedad hepática activa. Edad avanzada La eficacia y seguridad en pacientes mayores de 70 años, utilizando las dosis recomendadas, son similares a las observadas en la población general. Población pediátrica Hipercolesterolemia: El uso en pediatría solo se debe realizar por médicos con experiencia en el tratamiento de la hiperlipidemia pediátrica y los pacientes deben ser re-evaluados de forma periódica para verificar su progreso. La dosis inicial recomendada de atorvastatina, en pacientes con hipercolesterolemia familiar heterocigótica a partir de los 10 años, es de 10 mg al día. La dosis se puede aumentar hasta 80 mg al día, de acuerdo con la respuesta y la tolerabilidad. Las dosis se deben individualizar de acuerdo con el objetivo recomendado del tratamiento. Los ajustes de dosis se deben realizar a intervalos de 4 o más semanas. El ajuste de la dosis hasta 80 mg al día está respaldado por los datos de estudios en adultos y por los datos clínicos limitados de estudios en niños con hipercolesterolemia familiar heterocigótica. La dosis inicial recomendada de Atorvastatina, en pacientes con hipercolesterolemia familiar heterocigótica mayores de 6 años pero menores de 10 años, es de 5 a 10 mg al día.
CONTRAINDICACIONES: hipersensibilidad a alguno de sus componentes enfermedad hepática activa o elevación persistente de las transaminasas séricas (más de tres veces el límite normal superior) embarazo y lactancia. Advertencias y precauciones: efectos hepáticos se recomienda realizar pruebas de función hepática antes de iniciar el tratamiento y posteriormente de forma periódica. Se deben realizar pruebas de función hepática a los pacientes que desarrollen cualquier síntoma o signo que sugiera lesión hepática. Los pacientes que presenten un aumento en los niveles de transaminasas se deben controlar hasta que esta anomalía(s) quede(n) resuelta(s). En caso de un aumento persistente de las transaminasas 3 veces el valor máximo de normalidad, se recomienda una reducción de la dosis o la retirada de atorvastatina. Atorvastatina debe utilizarse con precaución en pacientes que consuman cantidades importantes de alcohol y/o con antecedentes de enfermedad hepática. Prevención del ictus mediante una reducción intensiva de los niveles de colesterol. En un análisis post-hoc de los subtipos de ictus en pacientes sin enfermedad coronaria (EC) que habían padecido recientemente un ictus o un accidente isquémico transitorio (AIT), se observó que había una mayor incidencia de ictus hemorrágico en aquellos pacientes en tratamiento con atorvastatina 80 mg en comparación con placebo. Este incremento del riesgo se observó especialmente en pacientes con ictus hemorrágico previo o infarto lacunar en el momento de la inclusión en el estudio. Para pacientes con ictus hemorrágico previo o infarto lacunar, el balance beneficio riesgo de atorvastatina 80 mg es incierto, y se habrá de considerar cuidadosamente el potencial riesgo de ictus hemorrágico antes de iniciar el tratamiento efectos en el músculo esquelético. Atorvastatina, como otros inhibidores de la HMG-CoA reductasa, puede afectar en raras ocasiones al músculo esquelético y producir mialgia, miositis y miopatía que pueden progresar a rabdomiólisis, una patología potencialmente mortal caracterizada por elevados niveles de creatina quinasa (CK) (> 10 veces el valor máximo de normalidad), mioglobinemia y mioglobinuria que puede producir insuficiencia renal. Se han notificado, en muy raras ocasiones, casos de miopatía necrotizante inmunomediada (MNIM) durante o después del tratamiento con algunas estatinas. Clínicamente, la MNIM se caracteriza por debilidad muscular proximal persistente y elevación de la creatina cinasa sérica, que persisten a pesar de la suspensión del tratamiento con la estatina. Antes de comenzar el tratamiento. Atorvastatina se debe prescribir con precaución en aquellos pacientes con factores que pueden predisponer a la aparición de rabdomiólisis. Antes de comenzar el tratamiento con estatinas, se deben determinar los niveles de CK en las siguientes situaciones: insuficiencia renal; hipotiroidismo. antecedentes personales o familiares de enfermedades musculares hereditarias. antecedentes de toxicidad muscular por una estatina o un fibrato; antecedentes de enfermedad hepática y/o cuando se consuman cantidades substanciales de alcohol. en ancianos (mayores de 70 años), la necesidad de estas determinaciones se debería valorar dependiendo de la existencia de otros factores predisponentes para el desarrollo de rabdomiólisis. situaciones en las que se puede producir un aumento en los niveles plasmáticos, como interacciones y en poblaciones especiales, incluyendo subpoblaciones genéticas. En todas las circunstancias enumeradas anteriormente, debe valorarse el riesgo del tratamiento frente a su posible beneficio y, se recomienda la vigilancia clínica del paciente. Si inicialmente los niveles de CK se encuentran significativamente elevados (> 5 veces el valor máximo de normalidad), el tratamiento no debe instaurarse. Determinación de la creatina quinasa. Los niveles de creatina quinasa (CK) no se deben determinar después de realizar un ejercicio físico intenso o en presencia de una causa alternativa que pueda explicar un incremento de la CK, ya que esto dificulta la interpretación del resultado. Si inicialmente los valores de CK están significativamente elevados (> 5 veces el valor máximo de normalidad), la determinación deberá repetirse de 5 a 7 días más tarde para confirmar estos resultados. Durante el tratamiento. – debe indicarse a los pacientes que comuniquen rápidamente cualquier dolor, calambres o debilidad muscular, especialmente si se acompaña de fiebre y malestar – si estos síntomas se presentan en pacientes que están en tratamiento con atorvastatina, se deben determinar sus niveles de CK. Si estos niveles resultan significativamente elevados (> 5 veces el valor máximo de normalidad) el tratamiento se debe interrumpir – en los casos en los que los síntomas sean severos y supongan molestias diarias para el paciente, se debe valorar la interrupción del tratamiento, incluso aunque los niveles de CK se encuentren elevados 5 veces el valor máximo de normalidad. Si los síntomas desaparecen y los niveles de CK se normalizan, se puede considerar la reintroducción de atorvastatina o bien la de otra estatina alternativa, a dosis más bajas y bajo estrecha vigilancia del paciente – debe interrumpirse el tratamiento con atorvastatina, si se produce una elevación clínicamente significativa de los niveles de CK (> 10 veces el valor máximo de normalidad), o si se diagnostica o sospecha rabdomiólisis. Tratamiento concomitante con otros medicamentos el riesgo de rabdomiólisis aumenta cuando se administra de forma concomitante atorvastatina con ciertos medicamentos que pueden incrementar su concentración plasmática, como inhibidores potentes de la cyp3a4 o proteínas transportadoras (por ejemplo, ciclosporina, telitromicina, claritromicina, delavirdina, estiripentol, ketoconazol, voriconazol, itraconazol, posaconazol e inhibidores de la proteasa del VIH incluyendo ritonavir, lopinavir, atazanavir, indinavir, darunavir, etc). El riesgo de miopatía, también puede verse incrementado, por el uso concomitante de gemfibrozilo y otros derivados del ácido fíbrico, boceprevir, eritromicina, niacina, ezetimiba, telaprevir o tipranavir y ritonavir combinados. Se deben considerar, cuando sea posible, terapias alternativas (que no interaccionen), en lugar de estos medicamentos. En los casos en los que la administración conjunta de estos medicamentos con atorvastatina sea necesaria, debe valorarse con cuidado el beneficio y el riesgo. Durante el tratamiento con medicamentos que aumenten las concentraciones plasmáticas de atorvastatina, se recomienda una dosis máxima de atorvastatina más baja. Además, en el caso de potentes inhibidores de la cyp3a4, debe considerarse una dosis inicial de atorvastatina más baja y se recomienda el seguimiento clínico de estos pacientes. Atorvastatina no debe administrarse conjuntamente con las formulaciones sistémicas de ácido fusídico o dentro de los 7 días de la interrupción del tratamiento con ácido fusídico. En pacientes en los que se considere esencial el uso de ácido fusídico sistémico, el tratamiento con estatinas debe interrumpirse durante toda la duración del tratamiento con ácido fusídico. Se han notificado casos de rabdomiólisis (incluyendo algunos caso mortales) en pacientes que recibieron ácido fusídico y estatinas en combinación. Se debe advertir al paciente que acuda inmediatamente al médico si experimenta algún síntoma de debilidad muscular, dolor o sensibilidad. Población pediátrica. No se ha observado ningún efecto clínicamente significativo sobre el crecimiento y la madurez sexual en un estudio de 3 años basado en la evaluación de la madurez y el desarrollo general, la evaluación de la clasificación de Tanner y la medición de la altura y el peso. Enfermedad pulmonar intersticial. Excepcionalmente se han notificado con algunas estatinas casos de enfermedad pulmonar intersticial, especialmente con tratamientos de larga. Los síntomas pueden incluir disnea, tos improductiva y malestar general (fatiga, pérdida de peso y fiebre). Si se sospecha que un paciente ha desarrollado enfermedad pulmonar intersticial, el tratamiento con estatinas debe interrumpirse. Diabetes mellitus. Algunas evidencias sugieren que las estatinas aumentan la glucosa en sangre y en algunos pacientes en riesgo de desarrollo de diabetes, pueden producir niveles de hiperglucemia donde los cuidados de la diabetes son necesarios. Este riesgo, sin embargo, es compensado por la reducción del riesgo cardiovascular con estatinas, por tanto no deber ser una razón para interrumpir el tratamiento con estatinas. Los pacientes con riesgo deben ser controlados desde el punto de vista clínico y bioquímico de acuerdo a las guías nacionales. Fertilidad, embarazo y lactancia mujeres en edad fértil las mujeres en edad fértil deben utilizar las medidas anticonceptivas adecuadas durante el tratamiento. Embarazo. Atorvastatina está contraindicado durante el embarazo. No se ha establecido la seguridad en mujeres embarazadas. No se han realizado ensayos clínicos controlados con atorvastatina en mujeres embarazadas. Raramente se han recibido notificaciones de anomalías congénitas tras la exposición intrauterina de inhibidores de la HMG-CoA reductasa. Los estudios en animales han mostrado toxicidad sobre la reproducción. El tratamiento de la madre con atorvastatina puede reducir los niveles fetales de mevalonato que es un precursor en la biosíntesis del colesterol. La aterosclerosis es un proceso crónico y normalmente la interrupción del tratamiento hipolipemiante durante el embarazo debe tener poco impacto sobre el riesgo a largo plazo asociado con la hipercolesterolemia primaria. Por esta razón, no se debe utilizar atorvastatina en mujeres embarazadas, que intentan quedarse embarazadas o sospechan que pudieran estarlo. El tratamiento con atorvastatina debe suspenderse durante el embarazo o hasta que se determine que la mujer no está embarazada. Lactancia. Se desconoce si atorvastatina o sus metabolitos se excretan a través de la leche humana. En ratas, las concentraciones plasmáticas de atorvastatina y sus metabolitos activos eran similares a las encontradas en la leche. Debido a su potencial para causar graves reacciones adversas, las mujeres que reciban atorvastatina no deben amamantar a sus hijos. Atorvastatina está contraindicada durante la lactancia. Lactosa: este medicamento contiene lactosa. Los pacientes con problemas hereditarios raros de intolerancia a la galactosa, deficiencia de lactasa Lapp o malabsorción de glucosa-galactosa no deben tomar este medicamento.
INTERACCIONES FARMACOLÓGICAS: Atorvastatina se metaboliza por la vía del citocromo P450 3A4 (CYP3A4) y es sustrato de las proteínas transportadoras, como por ejemplo, el transportador hepático OATP1B1. La administración concomitante de medicamentos que son inhibidores de la CYP3A4 o de proteínas transportadoras puede producir niveles elevados de las concentraciones plasmáticas de atorvastatina y un aumento del riesgo de miopatía. El riesgo también puede estar aumentado por la administración concomitante de atorvastatina con otros medicamentos con potencial para inducir miopatía, como derivados del ácido fíbrico y ezetimiba. Inhibidores de la CYP3A4 Los inhibidores potentes de la CYP3A4 han demostrado que producen concentraciones notablemente elevadas de atorvastatina (ver Tabla 1 y la información específica a continuación). Debe evitarse en lo posible, la administración concomitante de inhibidores potentes de la CYP3A4 (por ejemplo, ciclosporina, telitromicina, claritromicina, delavirdina, estiropentol, ketoconazol, voriconazol, itraconazol, posaconazol e inhibidores de la proteasa del VIH incluyendo ritonavir, lopinavir, atazanavir, indinavir, darunavir, etc). En los casos que no pueda evitarse la administración concomitante de estos medicamentos con atorvastatina, se debe considerar el uso de dosis inicial y máxima inferiores de atorvastatina y se recomienda el adecuado seguimiento clínico del paciente. Los inhibidores moderados de la CYP3A4 (por ejemplo, eritromicina, diltiazem, verapamilo y fluconazol) pueden aumentar las concentraciones plasmáticas de atorvastatina. Se ha observado un aumento en el riesgo de miopatía con el uso de eritromicina en combinación con estatinas. No se han realizado estudios de interacción para evaluar los efectos de amiodarona o verapamilo sobre atorvastatina. Se sabe que tanto amiodarona como verapamilo inhiben la actividad de la CYP3A4 y que su administración concomitante con atorvastatina puede llevar a una mayor exposición a atorvastatina. Por tanto, debe considerarse una dosis máxima de atorvastatina más baja y se recomienda el adecuado seguimiento clínico del paciente cuando se usa con inhibidores moderados de la CYP3A4. Se recomienda el adecuado seguimiento clínico tras el inicio o tras un ajuste de dosis del inhibidor. Inductores de la CYP3A4 La administración conjunta de atorvastatina con inductores del citocromo P450 3A4 (por ejemplo, efavirenz, rifampicina, hierba de San Juan) puede reducir de forma variable las concentraciones plasmáticas de atorvastatina. Debido al mecanismo de interacción doble de la rifampicina, (inducción del citocromo P450 3A4 e inhibición del transportador OATP1B1 del hepatocito), se recomienda la administración simultánea de atorvastatina con rifampicina, ya que la administración de atorvastatina tras la administración de rifampicina se ha asociado con una reducción significativa de las concentraciones plasmáticas de atorvastatina. Sin embargo, se desconoce el efecto de rifampicina sobre las concentraciones de atorvastatina en los hepatocitos, no obstante, si no se puede evitar la administración concomitante, se debe monitorizar cuidadosamente la eficacia en los pacientes. Inhibidores de las proteínas transportadoras Los inhibidores de las proteínas transportadoras (por ejemplo, ciclosporina) pueden aumentar la exposición sistémica a atorvastatina. Se desconoce el efecto de la inhibición de los transportadores hepáticos sobre las concentraciones de atorvastatina en el hepatocito. Si su administración concomitante no puede evitarse, se recomienda la reducción de la dosis y el seguimiento clínico de la eficacia. Gemfibrozilo/derivados del ácido fíbrico El uso de fibratos se ha asociado ocasionalmente con acontecimientos relacionados con el músculo, incluyendo rabdomiolísis. El riesgo de estos acontecimientos puede aumentar con la administración concomitante de derivados del ácido fíbrico y atorvastatina. Si su administración concomitante no puede evitarse, debe utilizarse la dosis más baja posible de atorvastatina para alcanzar el objetivo terapéutico y debe monitorizarse adecuadamente al paciente. Ezetimiba El uso de ezetimiba en monoterapia se asocia con acontecimientos relacionados con el músculo, incluyendo rabdomiolisis. El riesgo de esos acontecimientos puede por tanto estar aumentado con el uso concomitante de ezetimiba y atorvastatina. Se recomienda una adecuada monitorización clínica de estos pacientes. Colestipol Las concentraciones plasmáticas de atorvastatina y sus metabolitos activos fueron inferiores (proporción de concentración de atorvastatina: 0,74) cuando colestipol se administró junto con atorvastatina. No obstante, los efectos sobre los lípidos fueron mayores cuando se administraron conjuntamente atorvastatina y colestipol que cuando los fármacos se administraron por separado. Ácido fusídico El riesgo de miopatía incluyendo rabdomiólisis puede aumentar tras la administración concomitante de ácido fusídico sistémico con estatinas. El mecanismo de esta interacción (tanto farmacodinámica como farmacocinética, o ambas) aún no se conoce. Se han notificado casos de rabdomiólisis (incluyendo algunos casos mortales) en los pacientes que reciben esta combinación. Si el tratamiento con ácido fusídico sistémico es necesario, el uso de atorvastatina se debe suspender durante toda la duración del tratamiento con ácido fusídico. Colchicina Aunque no se han realizado estudios de interacciones con atorvastatina y colchicina, se han notificado casos de miopatía con atorvastatina cuando se administró de forma conjunta con colchicina, por lo que debe procederse con suma cautela cuando se prescriba atorvastatina con colchicina. Efecto de atorvastatina sobre medicamentos concomitantes Digoxina. Cuando se administraron conjuntamente dosis múltiples de digoxina y 10 mg de atorvastatina, las concentraciones plasmáticas en el estado estacionario de digoxina aumentaron ligeramente. Los pacientes tratados con digoxina deben ser monitorizados de forma adecuada. Anticonceptivos orales. La administración conjunta de atorvastatina con anticonceptivos orales produjo un aumento de las concentraciones plasmáticas de noretindrona y etinil estradiol. Warfarina. En un ensayo clínico en pacientes que recibían tratamiento crónico con warfarina, la administración concomitante de 80 mg al día de atorvastatina con warfarina produjo una pequeña reducción de aproximadamente 1,7 segundos en el tiempo de protrombina durante los primeros 4 días de tratamiento, que volvió a la normalidad en 15 días de tratamiento con atorvastatina. Aunque solo se han notificado muy raros casos de interacciones clínicamente significativas con anticoagulantes, debe determinarse el tiempo de protrombina antes de iniciar el tratamiento con atorvastatina en pacientes que reciban anticoagulantes cumarínicos y con una frecuencia suficiente al inicio del tratamiento para asegurar que no se produce una alteración significativa del tiempo de protrombina. Una vez que se haya determinado el tiempo de protrombina, podrán monitorizarse los tiempos de protrombina a los intervalos normalmente recomendados para los pacientes que reciben anticoagulantes cumarínicos. Si se cambia la dosis o se interrumpe el tratamiento con atorvastatina, debe repetirse el mismo procedimiento. El tratamiento con atorvastatina no se ha asociado con hemorragias o cambios en el tiempo de protrombina en pacientes que no reciben anticoagulantes.
EFECTOS SECUNDARIOS: En base a los datos de los estudios clínicos y de la amplia experiencia post-comercialización, se presenta a continuación el perfil de reacciones adversas de atorvastatina. Se ordenan las frecuencias estimadas para reacciones adversas de acuerdo con el siguiente criterio: frecuente (=1/100, < 1/10), poco frecuente (=1/1.000, <1/100); rara (=1/10.000, <1/1.000); muy rara (=1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Infecciones e infestaciones: Frecuente: nasofaringitis. Trastornos de la sangre y del sistema linfático Raro: trombocitopenia. Trastornos del sistema inmunológico Frecuente: reacciones alérgicas. Muy raro: anafilaxia. Trastornos del metabolismo y de la nutrición Frecuente: hiperglucemia. Poco frecuente: hipoglucemia, ganancia de peso, anorexia. Trastornos psiquiátricos Poco frecuente: pesadillas, insomnio. Trastornos del sistema nervioso Frecuente: cefalea. Poco frecuente: mareos, parestesia, hipoestesia, disgeusia, amnesia. Raro: neuropatía periférica. Trastornos oculares Poco frecuente: visión borrosa. Rara: alteración visual. Trastornos del oído y del laberinto Poco frecuente: acúfenos. Muy raros: pérdida de audición Trastornos respiratorios, torácicos y mediastínicos: Frecuente: dolor faringolaríngeo, epistaxis. Trastornos gastrointestinales Frecuentes: estreñimiento, flatulencia, dispepsia, nauseas, diarrea. Poco frecuentes: vómitos, dolor abdominal superior e inferior, eructos, pancreatitis. Trastornos hepatobiliares Poco frecuente: hepatitis. Raras: colestasis. Muy raras: insuficiencia hepática. Trastornos de la piel y del tejido subcutáneo Frecuentes: erupción, picor Poco frecuente: urticaria, erupción cutánea, prurito, alopecia. Rara: edema angioneurótico, dermatitis bullosa incluyendo eritema multiforme, síndrome de Stevens-Johnson y necrolisis epidérmica tóxica. Trastornos musculoesqueléticos y del tejido conjuntivo Frecuentes: mialgia, artralgia, dolor en las extremidades, espasmos musculares, hinchazón en las articulaciones, dolor de espalda. Poco frecuente: dolor de cuello, fatiga muscular. Raras: miopatía, miositis, rabdomiólisis, tendinopatía a veces complicada con ruptura. Frecuencia no conocida: miopatía necrotizante inmunomediada. Trastornos del aparato reproductor y de la mama Muy raros: ginecomastia Trastornos generales y alteraciones en el lugar de administración Poco frecuentes: malestar, astenia, dolor torácico, edema periférico, fatiga, pirexia. Exploraciones complementarias Frecuente: test de función hepática anormal, niveles elevados de creatinaquinasa en sangre. Poco frecuente: test de glóbulos blancos en orina positivo. Como con otros inhibidores de la HMG-CoA reductasa, se han notificado elevaciones en los niveles de las transaminasas séricas en los pacientes que recibían atorvastatina. Estos cambios fueron normalmente leves, transitorios y no requirieron interrupción del tratamiento. En un 0,8% de los pacientes que recibían atorvastatina se produjeron elevaciones clínicamente importantes (>3 veces por encima del valor máximo de normalidad) de las transaminasas séricas. Estas elevaciones estuvieron relacionadas con la dosis y fueron reversibles en todos los pacientes. En ensayos clínicos, al igual que con otros inhibidores de la HMG-CoA reductasa, un 2,5% de los pacientes tratados con atorvastatina presentaron niveles elevados de creatinaquinasa en suero 3 veces superiores al máximo de normalidad. En un 0,4% de los pacientes tratados con atorvastatina se observaron incrementos en valores 10 veces superiores al límite máximo de normalidad. Población pediátrica La base de datos de seguridad clínica incluye datos de seguridad de 249 pacientes pediátricos que recibieron atorvastatina, de los cuales 7 eran < 6 años, 14 estaban en el intervalo entre 6 a 9 años y 228 pacientes estaban en el intervalo de 10 a 17 años. Trastornos en el sistema nervioso Frecuente: Cefalea Trastornos gastrointestinales Frecuente: Dolor abdominal. Exploraciones complementarias Frecuentes: Alanina aminotransferasa elevada, creatinfosfoquinasa elevada en sangre En base a los datos disponibles, se espera que la frecuencia, tipo y gravedad de las reacciones adversas en niños sea igual a la de los adultos. Actualmente, la experiencia sobre la seguridad a largo plazo en la población pediátrica es limitada. Efectos de clase:Disfunción sexual, Depresión. Casos excepcionales de enfermedad pulmonar intersticial, especialmente con tratamientos a largo plazo. Diabetes mellitus: La frecuencia dependerá de la presencia o ausencia de factores de riesgo (glucosa sanguínea en ayunas =5,6 mmol/l, IMC >30 kg/m2, aumento de triglicéridos, historial de hipertensión).
PROPIEDADES FARMACODINÁMICAS: Atorvastatina es un inhibidor selectivo y competitivo de la HMG-CoA reductasa, la enzima limitante responsable de la conversión del 3-hidroxi-3-metil-glutaril-coenzima A a mevalonato, un precursor de los esteroles, incluyendo el colesterol. Los triglicéridos y el colesterol se incorporan a las lipoproteínas de muy baja densidad (VLDL) en el hígado y se liberan al plasma para su distribución por los tejidos periféricos. Las lipoproteínas de baja densidad (LDL) se forman a partir de las VLDL y se catabolizan principalmente a través del receptor con elevada afinidad para las LDL (receptor LDL). Atorvastatina reduce las concentraciones plasmáticas de colesterol y de lipoproteínas inhibiendo en el hígado la HMG-CoA reductasa y la subsiguiente biosíntesis hepática de colesterol y aumentando el número de receptores hepáticos para la LDL en la superficie celular, lo que da lugar a un incremento de la absorción y el catabolismo de las LDL.
CONDICIÓN DE VENTA: Venta bajo fórmula médica
PRESENTACIÓN: Caja por 10, 30 tabletas recubiertas. Reg. San INVIMA 2023M-0020877.

Colestox®
REGISTRO SANITARIO:
INVIMA 2023M-0020877
PRINCIPIO ACTIVO:
Rosuvastatina
UNIDAD DE VENTA:
Tabletas
Inhibidores HMG CoA Reductasa
(Atorvastatina)
COMPOSICIÓN: Cada tableta recubierta contiene 40 mg de atorvastatina, excipientes c.s.
INDICACIONES: la atorvastatina está indicada como coadyuvante de la dieta en el manejo de las dislipoproteinemias. Útil en pacientes con múltiples factores de riesgo para enfermedad cardíaca coronaria, las cuales pueden incluir diabetes mellitus, historia de trombosis u otra enfermedad cerebrovascular o enfermedad cardíaca coronaria asintomática, para disminuir el riesgo de infarto de miocardio no fatal y trombosis no fatal. Atorvastatina también está indicada para la reducción del colesterol total y colesterol LDL en pacientes con hipercolesterolemia familiar homocigótica y heterocigótica. Reduce el riesgo de infarto del miocardio no fatal, eventos cerebrales fatales y no fatales, angina, procedimientos de revascularización y necesidad de hospitalización por insuficiencia cardíaca congestiva en adultos con enfermedad coronaria clínicamente evidente, con niveles de colesterol controlado. Está indicada en pacientes con enfermedad vascular periférica o enfermedad cardíaca coronaria asintomática para disminuir el riesgo de infarto al miocardio no fatal y trombosis no fatal. Uso pediátrico para niños mayores de 6 años.
VÍA DE ADMINISTRACIÓN: Oral
FORMA FARMACÉUTICA: tableta recubierta.
POSOLOGÍA Y DOSIFICACIÓN: La dosis inicial habitual es de 10 mg una vez al día.
El ajuste de la dosis se debe hacer a intervalos de 4 o más semanas. La dosis máxima es de 80 mg una vez al día. Hipercolesterolemia primaria e hiperlipidemia combinada (mixta) La mayoría de los pacientes se controlan con Atorvastatina 10 mg administrado una vez al día. La respuesta terapéutica se observa al cabo de 2 semanas y habitualmente se alcanza la máxima respuesta terapéutica a las 4 semanas. La respuesta se mantiene durante el tratamiento crónico. Hipercolesterolemia familiar heterocigótica Los pacientes deben iniciar el tratamiento con 10 mg de Atorvastatina al día. Las dosis deben individualizarse y ajustarse cada 4 semanas hasta los 40 mg al día. Posteriormente, la dosis puede aumentarse hasta un máximo de 80 mg al día o se puede combinar 40 mg de atorvastatina una vez al día con un secuestrante de ácidos biliares. Hipercolesterolemia familiar homocigótica: Sólo se dispone de datos limitados. La dosis de atorvastatina en pacientes con hipercolesterolemia familiar homocigótica es de 10 a 80 mg al día. Atorvastatina debe utilizarse en terapia combinada con otros tratamientos hipolipemiantes (por ejemplo, aféresis de las LDL) en estos pacientes o si no se dispone de estos tratamientos. Prevención de la enfermedad cardiovascular: en los estudios en prevención primaria la dosis fue 10 mg/día. Pueden ser necesarias dosis mayores a fin de alcanzar los niveles de colesterol LDL de acuerdo con las guías actuales. Insuficiencia renal No es necesario un ajuste de la dosis. Insuficiencia hepática: atorvastatina se debe utilizar con precaución en pacientes con insuficiencia hepática. Atorvastatina está contraindicado en pacientes con enfermedad hepática activa. Edad avanzada La eficacia y seguridad en pacientes mayores de 70 años, utilizando las dosis recomendadas, son similares a las observadas en la población general. Población pediátrica Hipercolesterolemia: El uso en pediatría solo se debe realizar por médicos con experiencia en el tratamiento de la hiperlipidemia pediátrica y los pacientes deben ser re-evaluados de forma periódica para verificar su progreso. La dosis inicial recomendada de atorvastatina, en pacientes con hipercolesterolemia familiar heterocigótica a partir de los 10 años, es de 10 mg al día. La dosis se puede aumentar hasta 80 mg al día, de acuerdo con la respuesta y la tolerabilidad. Las dosis se deben individualizar de acuerdo con el objetivo recomendado del tratamiento. Los ajustes de dosis se deben realizar a intervalos de 4 o más semanas. El ajuste de la dosis hasta 80 mg al día está respaldado por los datos de estudios en adultos y por los datos clínicos limitados de estudios en niños con hipercolesterolemia familiar heterocigótica. La dosis inicial recomendada de Atorvastatina, en pacientes con hipercolesterolemia familiar heterocigótica mayores de 6 años pero menores de 10 años, es de 5 a 10 mg al día.
CONTRAINDICACIONES: hipersensibilidad a alguno de sus componentes enfermedad hepática activa o elevación persistente de las transaminasas séricas (más de tres veces el límite normal superior) embarazo y lactancia. Advertencias y precauciones: efectos hepáticos se recomienda realizar pruebas de función hepática antes de iniciar el tratamiento y posteriormente de forma periódica. Se deben realizar pruebas de función hepática a los pacientes que desarrollen cualquier síntoma o signo que sugiera lesión hepática. Los pacientes que presenten un aumento en los niveles de transaminasas se deben controlar hasta que esta anomalía(s) quede(n) resuelta(s). En caso de un aumento persistente de las transaminasas 3 veces el valor máximo de normalidad, se recomienda una reducción de la dosis o la retirada de atorvastatina. Atorvastatina debe utilizarse con precaución en pacientes que consuman cantidades importantes de alcohol y/o con antecedentes de enfermedad hepática. Prevención del ictus mediante una reducción intensiva de los niveles de colesterol. En un análisis post-hoc de los subtipos de ictus en pacientes sin enfermedad coronaria (EC) que habían padecido recientemente un ictus o un accidente isquémico transitorio (AIT), se observó que había una mayor incidencia de ictus hemorrágico en aquellos pacientes en tratamiento con atorvastatina 80 mg en comparación con placebo. Este incremento del riesgo se observó especialmente en pacientes con ictus hemorrágico previo o infarto lacunar en el momento de la inclusión en el estudio. Para pacientes con ictus hemorrágico previo o infarto lacunar, el balance beneficio riesgo de atorvastatina 80 mg es incierto, y se habrá de considerar cuidadosamente el potencial riesgo de ictus hemorrágico antes de iniciar el tratamiento efectos en el músculo esquelético. Atorvastatina, como otros inhibidores de la HMG-CoA reductasa, puede afectar en raras ocasiones al músculo esquelético y producir mialgia, miositis y miopatía que pueden progresar a rabdomiólisis, una patología potencialmente mortal caracterizada por elevados niveles de creatina quinasa (CK) (> 10 veces el valor máximo de normalidad), mioglobinemia y mioglobinuria que puede producir insuficiencia renal. Se han notificado, en muy raras ocasiones, casos de miopatía necrotizante inmunomediada (MNIM) durante o después del tratamiento con algunas estatinas. Clínicamente, la MNIM se caracteriza por debilidad muscular proximal persistente y elevación de la creatina cinasa sérica, que persisten a pesar de la suspensión del tratamiento con la estatina. Antes de comenzar el tratamiento. Atorvastatina se debe prescribir con precaución en aquellos pacientes con factores que pueden predisponer a la aparición de rabdomiólisis. Antes de comenzar el tratamiento con estatinas, se deben determinar los niveles de CK en las siguientes situaciones: insuficiencia renal; hipotiroidismo. antecedentes personales o familiares de enfermedades musculares hereditarias. antecedentes de toxicidad muscular por una estatina o un fibrato; antecedentes de enfermedad hepática y/o cuando se consuman cantidades substanciales de alcohol. en ancianos (mayores de 70 años), la necesidad de estas determinaciones se debería valorar dependiendo de la existencia de otros factores predisponentes para el desarrollo de rabdomiólisis. situaciones en las que se puede producir un aumento en los niveles plasmáticos, como interacciones y en poblaciones especiales, incluyendo subpoblaciones genéticas. En todas las circunstancias enumeradas anteriormente, debe valorarse el riesgo del tratamiento frente a su posible beneficio y, se recomienda la vigilancia clínica del paciente. Si inicialmente los niveles de CK se encuentran significativamente elevados (> 5 veces el valor máximo de normalidad), el tratamiento no debe instaurarse. Determinación de la creatina quinasa. Los niveles de creatina quinasa (CK) no se deben determinar después de realizar un ejercicio físico intenso o en presencia de una causa alternativa que pueda explicar un incremento de la CK, ya que esto dificulta la interpretación del resultado. Si inicialmente los valores de CK están significativamente elevados (> 5 veces el valor máximo de normalidad), la determinación deberá repetirse de 5 a 7 días más tarde para confirmar estos resultados. Durante el tratamiento. – debe indicarse a los pacientes que comuniquen rápidamente cualquier dolor, calambres o debilidad muscular, especialmente si se acompaña de fiebre y malestar – si estos síntomas se presentan en pacientes que están en tratamiento con atorvastatina, se deben determinar sus niveles de CK. Si estos niveles resultan significativamente elevados (> 5 veces el valor máximo de normalidad) el tratamiento se debe interrumpir – en los casos en los que los síntomas sean severos y supongan molestias diarias para el paciente, se debe valorar la interrupción del tratamiento, incluso aunque los niveles de CK se encuentren elevados 5 veces el valor máximo de normalidad. Si los síntomas desaparecen y los niveles de CK se normalizan, se puede considerar la reintroducción de atorvastatina o bien la de otra estatina alternativa, a dosis más bajas y bajo estrecha vigilancia del paciente – debe interrumpirse el tratamiento con atorvastatina, si se produce una elevación clínicamente significativa de los niveles de CK (> 10 veces el valor máximo de normalidad), o si se diagnostica o sospecha rabdomiólisis. Tratamiento concomitante con otros medicamentos el riesgo de rabdomiólisis aumenta cuando se administra de forma concomitante atorvastatina con ciertos medicamentos que pueden incrementar su concentración plasmática, como inhibidores potentes de la cyp3a4 o proteínas transportadoras (por ejemplo, ciclosporina, telitromicina, claritromicina, delavirdina, estiripentol, ketoconazol, voriconazol, itraconazol, posaconazol e inhibidores de la proteasa del VIH incluyendo ritonavir, lopinavir, atazanavir, indinavir, darunavir, etc). El riesgo de miopatía, también puede verse incrementado, por el uso concomitante de gemfibrozilo y otros derivados del ácido fíbrico, boceprevir, eritromicina, niacina, ezetimiba, telaprevir o tipranavir y ritonavir combinados. Se deben considerar, cuando sea posible, terapias alternativas (que no interaccionen), en lugar de estos medicamentos. En los casos en los que la administración conjunta de estos medicamentos con atorvastatina sea necesaria, debe valorarse con cuidado el beneficio y el riesgo. Durante el tratamiento con medicamentos que aumenten las concentraciones plasmáticas de atorvastatina, se recomienda una dosis máxima de atorvastatina más baja. Además, en el caso de potentes inhibidores de la cyp3a4, debe considerarse una dosis inicial de atorvastatina más baja y se recomienda el seguimiento clínico de estos pacientes. Atorvastatina no debe administrarse conjuntamente con las formulaciones sistémicas de ácido fusídico o dentro de los 7 días de la interrupción del tratamiento con ácido fusídico. En pacientes en los que se considere esencial el uso de ácido fusídico sistémico, el tratamiento con estatinas debe interrumpirse durante toda la duración del tratamiento con ácido fusídico. Se han notificado casos de rabdomiólisis (incluyendo algunos caso mortales) en pacientes que recibieron ácido fusídico y estatinas en combinación. Se debe advertir al paciente que acuda inmediatamente al médico si experimenta algún síntoma de debilidad muscular, dolor o sensibilidad. Población pediátrica. No se ha observado ningún efecto clínicamente significativo sobre el crecimiento y la madurez sexual en un estudio de 3 años basado en la evaluación de la madurez y el desarrollo general, la evaluación de la clasificación de Tanner y la medición de la altura y el peso. Enfermedad pulmonar intersticial. Excepcionalmente se han notificado con algunas estatinas casos de enfermedad pulmonar intersticial, especialmente con tratamientos de larga. Los síntomas pueden incluir disnea, tos improductiva y malestar general (fatiga, pérdida de peso y fiebre). Si se sospecha que un paciente ha desarrollado enfermedad pulmonar intersticial, el tratamiento con estatinas debe interrumpirse. Diabetes mellitus. Algunas evidencias sugieren que las estatinas aumentan la glucosa en sangre y en algunos pacientes en riesgo de desarrollo de diabetes, pueden producir niveles de hiperglucemia donde los cuidados de la diabetes son necesarios. Este riesgo, sin embargo, es compensado por la reducción del riesgo cardiovascular con estatinas, por tanto no deber ser una razón para interrumpir el tratamiento con estatinas. Los pacientes con riesgo deben ser controlados desde el punto de vista clínico y bioquímico de acuerdo a las guías nacionales. Fertilidad, embarazo y lactancia mujeres en edad fértil las mujeres en edad fértil deben utilizar las medidas anticonceptivas adecuadas durante el tratamiento. Embarazo. Atorvastatina está contraindicado durante el embarazo. No se ha establecido la seguridad en mujeres embarazadas. No se han realizado ensayos clínicos controlados con atorvastatina en mujeres embarazadas. Raramente se han recibido notificaciones de anomalías congénitas tras la exposición intrauterina de inhibidores de la HMG-CoA reductasa. Los estudios en animales han mostrado toxicidad sobre la reproducción. El tratamiento de la madre con atorvastatina puede reducir los niveles fetales de mevalonato que es un precursor en la biosíntesis del colesterol. La aterosclerosis es un proceso crónico y normalmente la interrupción del tratamiento hipolipemiante durante el embarazo debe tener poco impacto sobre el riesgo a largo plazo asociado con la hipercolesterolemia primaria. Por esta razón, no se debe utilizar atorvastatina en mujeres embarazadas, que intentan quedarse embarazadas o sospechan que pudieran estarlo. El tratamiento con atorvastatina debe suspenderse durante el embarazo o hasta que se determine que la mujer no está embarazada. Lactancia. Se desconoce si atorvastatina o sus metabolitos se excretan a través de la leche humana. En ratas, las concentraciones plasmáticas de atorvastatina y sus metabolitos activos eran similares a las encontradas en la leche. Debido a su potencial para causar graves reacciones adversas, las mujeres que reciban atorvastatina no deben amamantar a sus hijos. Atorvastatina está contraindicada durante la lactancia. Lactosa: este medicamento contiene lactosa. Los pacientes con problemas hereditarios raros de intolerancia a la galactosa, deficiencia de lactasa Lapp o malabsorción de glucosa-galactosa no deben tomar este medicamento.
INTERACCIONES FARMACOLÓGICAS: Atorvastatina se metaboliza por la vía del citocromo P450 3A4 (CYP3A4) y es sustrato de las proteínas transportadoras, como por ejemplo, el transportador hepático OATP1B1. La administración concomitante de medicamentos que son inhibidores de la CYP3A4 o de proteínas transportadoras puede producir niveles elevados de las concentraciones plasmáticas de atorvastatina y un aumento del riesgo de miopatía. El riesgo también puede estar aumentado por la administración concomitante de atorvastatina con otros medicamentos con potencial para inducir miopatía, como derivados del ácido fíbrico y ezetimiba. Inhibidores de la CYP3A4 Los inhibidores potentes de la CYP3A4 han demostrado que producen concentraciones notablemente elevadas de atorvastatina (ver Tabla 1 y la información específica a continuación). Debe evitarse en lo posible, la administración concomitante de inhibidores potentes de la CYP3A4 (por ejemplo, ciclosporina, telitromicina, claritromicina, delavirdina, estiropentol, ketoconazol, voriconazol, itraconazol, posaconazol e inhibidores de la proteasa del VIH incluyendo ritonavir, lopinavir, atazanavir, indinavir, darunavir, etc). En los casos que no pueda evitarse la administración concomitante de estos medicamentos con atorvastatina, se debe considerar el uso de dosis inicial y máxima inferiores de atorvastatina y se recomienda el adecuado seguimiento clínico del paciente. Los inhibidores moderados de la CYP3A4 (por ejemplo, eritromicina, diltiazem, verapamilo y fluconazol) pueden aumentar las concentraciones plasmáticas de atorvastatina. Se ha observado un aumento en el riesgo de miopatía con el uso de eritromicina en combinación con estatinas. No se han realizado estudios de interacción para evaluar los efectos de amiodarona o verapamilo sobre atorvastatina. Se sabe que tanto amiodarona como verapamilo inhiben la actividad de la CYP3A4 y que su administración concomitante con atorvastatina puede llevar a una mayor exposición a atorvastatina. Por tanto, debe considerarse una dosis máxima de atorvastatina más baja y se recomienda el adecuado seguimiento clínico del paciente cuando se usa con inhibidores moderados de la CYP3A4. Se recomienda el adecuado seguimiento clínico tras el inicio o tras un ajuste de dosis del inhibidor. Inductores de la CYP3A4 La administración conjunta de atorvastatina con inductores del citocromo P450 3A4 (por ejemplo, efavirenz, rifampicina, hierba de San Juan) puede reducir de forma variable las concentraciones plasmáticas de atorvastatina. Debido al mecanismo de interacción doble de la rifampicina, (inducción del citocromo P450 3A4 e inhibición del transportador OATP1B1 del hepatocito), se recomienda la administración simultánea de atorvastatina con rifampicina, ya que la administración de atorvastatina tras la administración de rifampicina se ha asociado con una reducción significativa de las concentraciones plasmáticas de atorvastatina. Sin embargo, se desconoce el efecto de rifampicina sobre las concentraciones de atorvastatina en los hepatocitos, no obstante, si no se puede evitar la administración concomitante, se debe monitorizar cuidadosamente la eficacia en los pacientes. Inhibidores de las proteínas transportadoras Los inhibidores de las proteínas transportadoras (por ejemplo, ciclosporina) pueden aumentar la exposición sistémica a atorvastatina. Se desconoce el efecto de la inhibición de los transportadores hepáticos sobre las concentraciones de atorvastatina en el hepatocito. Si su administración concomitante no puede evitarse, se recomienda la reducción de la dosis y el seguimiento clínico de la eficacia. Gemfibrozilo/derivados del ácido fíbrico El uso de fibratos se ha asociado ocasionalmente con acontecimientos relacionados con el músculo, incluyendo rabdomiolísis. El riesgo de estos acontecimientos puede aumentar con la administración concomitante de derivados del ácido fíbrico y atorvastatina. Si su administración concomitante no puede evitarse, debe utilizarse la dosis más baja posible de atorvastatina para alcanzar el objetivo terapéutico y debe monitorizarse adecuadamente al paciente. Ezetimiba El uso de ezetimiba en monoterapia se asocia con acontecimientos relacionados con el músculo, incluyendo rabdomiolisis. El riesgo de esos acontecimientos puede por tanto estar aumentado con el uso concomitante de ezetimiba y atorvastatina. Se recomienda una adecuada monitorización clínica de estos pacientes. Colestipol Las concentraciones plasmáticas de atorvastatina y sus metabolitos activos fueron inferiores (proporción de concentración de atorvastatina: 0,74) cuando colestipol se administró junto con atorvastatina. No obstante, los efectos sobre los lípidos fueron mayores cuando se administraron conjuntamente atorvastatina y colestipol que cuando los fármacos se administraron por separado. Ácido fusídico El riesgo de miopatía incluyendo rabdomiólisis puede aumentar tras la administración concomitante de ácido fusídico sistémico con estatinas. El mecanismo de esta interacción (tanto farmacodinámica como farmacocinética, o ambas) aún no se conoce. Se han notificado casos de rabdomiólisis (incluyendo algunos casos mortales) en los pacientes que reciben esta combinación. Si el tratamiento con ácido fusídico sistémico es necesario, el uso de atorvastatina se debe suspender durante toda la duración del tratamiento con ácido fusídico. Colchicina Aunque no se han realizado estudios de interacciones con atorvastatina y colchicina, se han notificado casos de miopatía con atorvastatina cuando se administró de forma conjunta con colchicina, por lo que debe procederse con suma cautela cuando se prescriba atorvastatina con colchicina. Efecto de atorvastatina sobre medicamentos concomitantes Digoxina. Cuando se administraron conjuntamente dosis múltiples de digoxina y 10 mg de atorvastatina, las concentraciones plasmáticas en el estado estacionario de digoxina aumentaron ligeramente. Los pacientes tratados con digoxina deben ser monitorizados de forma adecuada. Anticonceptivos orales. La administración conjunta de atorvastatina con anticonceptivos orales produjo un aumento de las concentraciones plasmáticas de noretindrona y etinil estradiol. Warfarina. En un ensayo clínico en pacientes que recibían tratamiento crónico con warfarina, la administración concomitante de 80 mg al día de atorvastatina con warfarina produjo una pequeña reducción de aproximadamente 1,7 segundos en el tiempo de protrombina durante los primeros 4 días de tratamiento, que volvió a la normalidad en 15 días de tratamiento con atorvastatina. Aunque solo se han notificado muy raros casos de interacciones clínicamente significativas con anticoagulantes, debe determinarse el tiempo de protrombina antes de iniciar el tratamiento con atorvastatina en pacientes que reciban anticoagulantes cumarínicos y con una frecuencia suficiente al inicio del tratamiento para asegurar que no se produce una alteración significativa del tiempo de protrombina. Una vez que se haya determinado el tiempo de protrombina, podrán monitorizarse los tiempos de protrombina a los intervalos normalmente recomendados para los pacientes que reciben anticoagulantes cumarínicos. Si se cambia la dosis o se interrumpe el tratamiento con atorvastatina, debe repetirse el mismo procedimiento. El tratamiento con atorvastatina no se ha asociado con hemorragias o cambios en el tiempo de protrombina en pacientes que no reciben anticoagulantes.
EFECTOS SECUNDARIOS: En base a los datos de los estudios clínicos y de la amplia experiencia post-comercialización, se presenta a continuación el perfil de reacciones adversas de atorvastatina. Se ordenan las frecuencias estimadas para reacciones adversas de acuerdo con el siguiente criterio: frecuente (=1/100, < 1/10), poco frecuente (=1/1.000, <1/100); rara (=1/10.000, <1/1.000); muy rara (=1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Infecciones e infestaciones: Frecuente: nasofaringitis. Trastornos de la sangre y del sistema linfático Raro: trombocitopenia. Trastornos del sistema inmunológico Frecuente: reacciones alérgicas. Muy raro: anafilaxia. Trastornos del metabolismo y de la nutrición Frecuente: hiperglucemia. Poco frecuente: hipoglucemia, ganancia de peso, anorexia. Trastornos psiquiátricos Poco frecuente: pesadillas, insomnio. Trastornos del sistema nervioso Frecuente: cefalea. Poco frecuente: mareos, parestesia, hipoestesia, disgeusia, amnesia. Raro: neuropatía periférica. Trastornos oculares Poco frecuente: visión borrosa. Rara: alteración visual. Trastornos del oído y del laberinto Poco frecuente: acúfenos. Muy raros: pérdida de audición Trastornos respiratorios, torácicos y mediastínicos: Frecuente: dolor faringolaríngeo, epistaxis. Trastornos gastrointestinales Frecuentes: estreñimiento, flatulencia, dispepsia, nauseas, diarrea. Poco frecuentes: vómitos, dolor abdominal superior e inferior, eructos, pancreatitis. Trastornos hepatobiliares Poco frecuente: hepatitis. Raras: colestasis. Muy raras: insuficiencia hepática. Trastornos de la piel y del tejido subcutáneo Frecuentes: erupción, picor Poco frecuente: urticaria, erupción cutánea, prurito, alopecia. Rara: edema angioneurótico, dermatitis bullosa incluyendo eritema multiforme, síndrome de Stevens-Johnson y necrolisis epidérmica tóxica. Trastornos musculoesqueléticos y del tejido conjuntivo Frecuentes: mialgia, artralgia, dolor en las extremidades, espasmos musculares, hinchazón en las articulaciones, dolor de espalda. Poco frecuente: dolor de cuello, fatiga muscular. Raras: miopatía, miositis, rabdomiólisis, tendinopatía a veces complicada con ruptura. Frecuencia no conocida: miopatía necrotizante inmunomediada. Trastornos del aparato reproductor y de la mama Muy raros: ginecomastia Trastornos generales y alteraciones en el lugar de administración Poco frecuentes: malestar, astenia, dolor torácico, edema periférico, fatiga, pirexia. Exploraciones complementarias Frecuente: test de función hepática anormal, niveles elevados de creatinaquinasa en sangre. Poco frecuente: test de glóbulos blancos en orina positivo. Como con otros inhibidores de la HMG-CoA reductasa, se han notificado elevaciones en los niveles de las transaminasas séricas en los pacientes que recibían atorvastatina. Estos cambios fueron normalmente leves, transitorios y no requirieron interrupción del tratamiento. En un 0,8% de los pacientes que recibían atorvastatina se produjeron elevaciones clínicamente importantes (>3 veces por encima del valor máximo de normalidad) de las transaminasas séricas. Estas elevaciones estuvieron relacionadas con la dosis y fueron reversibles en todos los pacientes. En ensayos clínicos, al igual que con otros inhibidores de la HMG-CoA reductasa, un 2,5% de los pacientes tratados con atorvastatina presentaron niveles elevados de creatinaquinasa en suero 3 veces superiores al máximo de normalidad. En un 0,4% de los pacientes tratados con atorvastatina se observaron incrementos en valores 10 veces superiores al límite máximo de normalidad. Población pediátrica La base de datos de seguridad clínica incluye datos de seguridad de 249 pacientes pediátricos que recibieron atorvastatina, de los cuales 7 eran < 6 años, 14 estaban en el intervalo entre 6 a 9 años y 228 pacientes estaban en el intervalo de 10 a 17 años. Trastornos en el sistema nervioso Frecuente: Cefalea Trastornos gastrointestinales Frecuente: Dolor abdominal. Exploraciones complementarias Frecuentes: Alanina aminotransferasa elevada, creatinfosfoquinasa elevada en sangre En base a los datos disponibles, se espera que la frecuencia, tipo y gravedad de las reacciones adversas en niños sea igual a la de los adultos. Actualmente, la experiencia sobre la seguridad a largo plazo en la población pediátrica es limitada. Efectos de clase:Disfunción sexual, Depresión. Casos excepcionales de enfermedad pulmonar intersticial, especialmente con tratamientos a largo plazo. Diabetes mellitus: La frecuencia dependerá de la presencia o ausencia de factores de riesgo (glucosa sanguínea en ayunas =5,6 mmol/l, IMC >30 kg/m2, aumento de triglicéridos, historial de hipertensión).
PROPIEDADES FARMACODINÁMICAS: Atorvastatina es un inhibidor selectivo y competitivo de la HMG-CoA reductasa, la enzima limitante responsable de la conversión del 3-hidroxi-3-metil-glutaril-coenzima A a mevalonato, un precursor de los esteroles, incluyendo el colesterol. Los triglicéridos y el colesterol se incorporan a las lipoproteínas de muy baja densidad (VLDL) en el hígado y se liberan al plasma para su distribución por los tejidos periféricos. Las lipoproteínas de baja densidad (LDL) se forman a partir de las VLDL y se catabolizan principalmente a través del receptor con elevada afinidad para las LDL (receptor LDL). Atorvastatina reduce las concentraciones plasmáticas de colesterol y de lipoproteínas inhibiendo en el hígado la HMG-CoA reductasa y la subsiguiente biosíntesis hepática de colesterol y aumentando el número de receptores hepáticos para la LDL en la superficie celular, lo que da lugar a un incremento de la absorción y el catabolismo de las LDL.
CONDICIÓN DE VENTA: Venta bajo fórmula médica
PRESENTACIÓN: Caja por 10, 30 tabletas recubiertas. Reg. San INVIMA 2023M-0020877.

Ezetimiba
REGISTRO SANITARIO:
INVIMA 2024-0021462
PRINCIPIO ACTIVO:
Ezetimiba
UNIDAD DE VENTA:
Tabletas
COMPOSICIÓN: Cada tableta contiene 10 mg de ezetimiba, excipientes c.s.
INDICACIONES: Hipercolesterolemia primaria. En combinación con fenofibrato, como terapia adyuvante a la dieta para la reducción de los niveles elevados del colesterol total (c total), el colesterol LDL (C-LDL), la apoliproteína b (APO B) y el colesterol no HDL (no C-HDL) en pacientes con hiperlipidemia mixta. Hipercolesterolemia familiar homocigótica (HOFH). Sitosterolemia homocigótica (fitosterolemia). Asociado a estatinas prevención secundaria de enfermedad cardiovascular en pacientes con enfermedad coronaria en cuanto a la indicación “prevención de eventos cardiovasculares mayores en enfermedad renal crónica” la sala considera que la evidencia allegada no es suficiente para determinar la verdadera utilidad y la relación riesgo/beneficio del producto de la referencia en esta indicación, teniendo en cuenta los resultados obtenidos en los estudios presentados y los criterios de inclusión de los pacientes en el estudio.
VÍA DE ADMINISTRACIÓN: Oral
FORMA FARMACÉUTICA: tableta.
DOSIFICACIÓN Y POSOLOGÍA: El paciente deberá seguir una dieta hipolipemiante adecuada, con la que debería continuar durante el tratamiento con ezetimiba. La dosis recomendada es un comprimido de 10 mg al día. Ezetimiba puede administrarse a cualquier hora del día, con o sin alimentos. Cuando la ezetimiba se añade a una estatina, debe continuarse con la administración de la estatina a la dosis de inicio habitual de la misma, o bien continuar con la dosis más alta previamente establecida. Ezetimiba deberá tomarse al menos 2 horas antes ó 4 horas después de la administración de un secuestrante de ácidos biliares. En cualquier caso, deben consultarse las instrucciones posológicas de la estatina. Uso en pacientes con cardiopatía coronaria y antecedentes de un acontecimiento de SCA. Para conseguir una reducción incremental de los eventos cardiovasculares en pacientes con cardiopatía coronaria y antecedentes de un acontecimiento de SCA, ezetimiba 10 mg puede administrarse con una estatina que tenga un beneficio cardiovascular demostrado. Administración con secuestrantes de ácidos biliares: ezetimiba deberá tomarse al menos 2 horas antes ó 4 horas después de la administración de un secuestrante de ácidos biliares. Pacientes de edad avanzada: No se precisa el ajuste de dosis en pacientes de edad avanzada. Población pediátrica: El inicio del tratamiento debe realizarse bajo la supervisión de un especialista. Niños y adolescentes de 6 años o mayores: No se ha establecido la seguridad y eficacia de ezetimiba en niños de 6 a 17 años. Cuando ezetimiba se administra junto con una estatina en niños, deben consultarse las instrucciones posológicas de la estatina. Niños menores de 6 años: No se ha establecido la seguridad y eficacia de ezetimiba en niños menores de 6 años. No se dispone de datos. Insuficiencia hepática: No es necesario ajustar la dosis de ezetimiba en pacientes con insuficiencia hepática leve (puntuación de Child-Pugh de 5 a 6). No se recomienda el tratamiento con ezetimiba en pacientes con disfunción hepática moderada (puntuación de Child-Pugh de 7 a 9) o grave (puntuación de Child-Pugh > 9). Insuficiencia renal: No es necesario ajustar la dosis en pacientes con insuficiencia renal.
CONTRAINDICACIONES: Hipersensibilidad a cualquier componente del medicamento. Embarazo y lactancia. Insuficiencia hepática moderada o severa. Niños menores de 10 años. ADVERTENCIAS Y PRECAUCIONES: si se administra con una estatina o un fenofibrato, por favor referirse al inserto de ese medicamento en particular. Su uso concomitante con estatinas requiere evaluación hepática. Reportar al médico cualquier dolor, sensibilidad o debilidad muscular inexplicable. No se recomienda su coadministración con fibratos diferentes al fenofibrato ni con otros secuestrantes como la colestiramina o colestipol. Se deben realizar monitoreos cuando se administra en pacientes que reciben ciclosporina o anticoagulantes.
INTERACCIONES: En los estudios preclínicos se ha demostrado que ezetimiba no induce las enzimas metabolizadoras de fármacos del sistema del citocromo P450, esto presenta un bajo potencial de interacciones con otros medicamentos. No se han observado interacciones farmacocinéticas clínicamente importantes entre ezetimiba y fármacos metabolizados por las isoenzimas 1A2, 2D6, 2C8, 2C9 y 3A4 del citocromo P450 o por la N-acetiltransferasa. En estudios de interacción clínica, ezetimiba no tuvo efecto sobre la farmacocinética de dapsona, dextrometorfano, digoxina, anticonceptivos orales (etinil estradiol y levonorgestrel), glipizida, tolbutamida o midazolam en su administración concomitante. Cimetidina no afectó la biodisponibilidad de ezetimiba cuando se administraron concomitantemente. Antiácidos La administración simultánea de antiácidos redujo la tasa de absorción de ezetimiba, pero no tuvo efecto sobre su biodisponibilidad. Esta reducción de la tasa de absorción no se consideró clínicamente relevante. Colestiramina: La administración simultánea de colestiramina redujo el valor medio del área bajo la curva (ABC) de ezetimiba total (ezetimiba + ezetimiba glucurónido) aproximadamente un 55 %. Es posible que el incremento en la reducción de C-LDL que se produciría al añadir ezetimiba a colestiramina, disminuya como consecuencia de esta interacción. Fibratos: en pacientes que están recibiendo ezetimiba y fenofibrato, los médicos deben conocer el posible riesgo de colelitiasis y enfermedad de la vesícula biliar. Si se sospecha colelitiasis en un paciente que está recibiendo ezetimiba y fenofibrato, están indicadas exploraciones de la vesícula biliar y este tratamiento deberá interrumpirse. La administración concomitante con fenofibrato o genfibrozilo aumenta la concentración total de ezetimiba (aproximadamente 1,5 y 1,7 veces respectivamente). Si se administra con secuestrantes de ácidos biliares (colestiramina, colestipol), deberá tomarse al menos 2 horas antes o 4 horas después de la administración de éstos. La administración simultánea reduce el valor medio de la concentración plasmática de ezetimiba aproximadamente en un 55%, por lo que es posible que el incremento en la reducción de cLDL que se debería producir al asociar ezetimiba y colestiramina no ocurra como consecuencia de esta interacción. Por otra parte, la administración concomitante con fibratos (fenofibrato, gemfibrozilo) aumenta la concentración total de ezetimiba entre 1,5 y 1,7 veces. Además, puede esperarse un incremento de la excreción de colesterol a la bilis, con aparición de colelitiasis. Los fibratos pueden incrementar la excreción del colesterol a la bilis y producir colelitiasis. En estudios en animales, algunas veces ezetimiba aumentó el colesterol en la vesícula biliar pero no en todas las especies. No puede excluirse un riesgo litogénico asociado con el uso terapéutico de ezetimiba. Estatinas: no se observaron interacciones farmacocinéticas clínicamente importantes cuando ezetimiba se administró junto con atorvastatina, simvastatina, pravastatina, lovastatina, fluvastatina o rosuvastatina. El uso concomitante de Rosuvastatina 10 mg con 10 mg de ezetimiba provocó un aumento de 1,2 veces en el AUC de rosuvastatina en sujetos hipercolesterolémicos. Sin embargo, no se puede descartar una interacción farmacodinámica, en términos de reacciones adversas, entre rosuvastatina y ezetimiba. Ciclosporina: la administración de una dosis única de 10 mg de ezetimiba a ocho pacientes sometidos a un trasplante renal con aclaramiento de creatinina superior a 50 ml/min y en tratamiento estable con ciclosporina, produjo un aumento de 3,4 veces (intervalo de 2,3 a 7,9 veces) del área bajo la curva (ABC) media para ezetimiba total en comparación con una población control sana, de otro estudio (n = 17) que estaba recibiendo únicamente ezetimiba. En un estudio diferente, se encontró en un paciente con trasplante renal y insuficiencia renal grave que recibía ciclosporina y otros medicamentos, una exposición a ezetimiba total 12 veces superior a la encontrada en sujetos control que estaban recibiendo únicamente ezetimiba. En un estudio cruzado de 2 periodos en doce sujetos sanos, la administración diaria de 20 mg de ezetimiba durante 8 días y una dosis única de 100 mg de ciclosporina el día 7, resultó en un aumento medio del 15% en el ABC de ciclosporina (intervalo del 10% de descenso al 51% de aumento), en comparación con los resultados obtenidos tras la administración de una dosis de 100 mg de ciclosporina sola. No se ha realizado un estudio controlado sobre el efecto de la administración conjunta de ezetimiba y ciclosporina sobre la exposición a ciclosporina en pacientes con trasplante renal. Se debe tener precaución al iniciar el tratamiento con ezetimiba en pacientes que reciben ciclosporina. Las concentraciones de ciclosporina deben vigilarse en pacientes que estén recibiendo ezetimiba y ciclosporina. Anticoagulantes: la administración concomitante de ezetimiba (10 mg una vez al día) no tuvo un efecto significativo sobre la biodisponibilidad de warfarina y el tiempo de protrombina en un estudio en doce varones adultos sanos. Sin embargo, se han recibido notificaciones después de la comercialización de aumento del cociente internacional normalizado (INR) en pacientes que tomaron ezetimiba con warfarina o fluindiona. Si se añade ezetimiba a warfarina, a otro anticoagulante cumarínico o a fluindiona, el INR debe ser vigilado apropiadamente.
EFECTOS SECUNDARIOS: Trastornos de la sangre y del sistema linfático No conocida: trombocitopenia. Trastornos del sistema inmunológico No conocida: hipersensibilidad; incluyendo erupción; urticaria; anafilaxia y angioedema. Trastornos del metabolismo y de la nutrición Poco frecuentes: apetito disminuido Trastornos psiquiátricos No conocida: depresión Trastornos del sistema nervioso Frecuentes cefalea Poco frecuentes: parestesia No conocida: mareo. Trastornos vasculares Poco frecuentes sofoco; hipertensión. Trastornos respiratorios, torácicos y mediastínicos Poco frecuentes: tos No conocida: disnea. Trastornos gastrointestinales Frecuentes: dolor abdominal; diarrea; flatulencia Poco frecuentes: dispepsia; enfermedad por reflujo gastroesofágico; náuseas; boca seca; gastritis No conocida: pancreatitis; estreñimiento. Trastornos hepatobiliares No conocida: hepatitis; colelitiasis; colecistitis. Trastornos de la piel y del tejido subcutáneo Poco frecuentes: prurito; erupción; urticaria No conocida: eritema multiforme. Trastornos musculoesqueléticos y del tejido conjuntivo Frecuentes: mialgia Poco frecuentes: artralgia; espasmos musculares; dolor de cuello; dolor de espalda; debilidad muscular; dolor en una extremidad. No conocida: miopatía/rabdomiólisis. Trastornos generales y alteraciones en el lugar de administración Frecuentes: fatiga. Poco frecuentes: dolor torácico; dolor; astenia; edema periférico. Exploraciones complementarias Frecuentes: ALT y/o AST elevadas. Poco frecuentes: CPK elevada en sangre; gamma glutamiltransferasa elevada; prueba de función hepática anormal.
INFORMACIÓN ADICIONAL: Considere realizar pruebas de función hepática antes de iniciar tratamiento y de manera rutinaria en pacientes en tratamiento y/o que vayan a iniciar, ya sea en monoterapia o en combinación con estatina o fibrato. De instrucciones a los pacientes respecto de consultar a un medico en caso de presentarse síntomas sugestivos de daño hepático inducido por medicamentos como dolor abdominal en flanco superior derecho, ictericia o ascitis. Recomiende a los pacientes suspender el tratamiento con ezetimiba si experimentan síntomas de reacción adversa cutánea como descamación de la piel, ampollas o pústulas en múltiples sectores del cuerpo (piel y mucosas), fiebre y síntomas parecidos a los de la gripa asociados, edema facial o de miembros, eritema multiforme o púrpura.
TOXICIDAD: En estudios clínicos, la administración de ezetimiba a 15 sujetos sanos a dosis de 50 mg/día durante 14 días, o 40 mg/día a 18 pacientes con hipercolesterolemia primaria durante 56 días, fue generalmente bien tolerada. En animales, no se observó toxicidad tras dosis orales únicas de 5.000 mg/kg de ezetimiba en la rata y el ratón y dosis de 3.000 mg/kg en el perro. Se han comunicado unos pocos casos de sobredosis con ezetimiba; la mayoría no se han asociado con experiencias adversas. Las experiencias adversas comunicadas no han sido graves. En caso de sobredosis, se deberán instituir medidas sintomáticas y de apoyo.
CLASIFICACIÓN EN EMBARAZO: Contraindicado en embarazo y lactancia.
PROPIEDADES FARMACODINÁMICAS: La diana molecular de ezetimiba es el transportador de esterol, el Niemann-Pick C1-Like 1 (NPC1L1), responsable de la captación intestinal de colesterol y fitoesteroles. Ezetimiba se localiza en las microvellosidades del intestino delgado e inhibe la absorción de colesterol, reduciendo el paso de colesterol desde el intestino al hígado; las estatinas reducen la síntesis de colesterol en el hígado y estos diferentes mecanismos juntos proporcionan una reducción complementaria del colesterol. En un ensayo clínico de 2 semanas de duración en el que se incluyeron 18 pacientes con hipercolesterolemia, ezetimiba inhibió la absorción intestinal de colesterol en un 54 % vs. placebo. Se realizaron una serie de estudios preclínicos para determinar la selectividad de ezetimiba para inhibir la absorción de colesterol. Ezetimiba inhibió la absorción de [14C]-colesterol sin mostrar efectos sobre la absorción de triglicéridos, ácidos grasos, ácidos biliares, progesterona, etinilestradiol o las vitaminas liposolubles A y D.
CONDICIÓN DE VENTA: Con fórmula facultativa.
PRESENTACIÓN: caja por 10 tabletas. Reg. San. INVIMA 2024-0021462

Prazed®
REGISTRO SANITARIO:
INVIMA 2019M-0013109-R1
PRINCIPIO ACTIVO:
Omeprazol
UNIDAD DE VENTA:
Cápsulas
Inhibidor de la bomba de
protones – antisecretor
gástrico – antiulceroso
(Omeprazol)
COMPOSICIÓN: Cada CÁPSULA contiene omeprazol 20 mg en microgránulos con cubierta entérica.
INDICACIONES: Úlcera péptica y duodenal, esofagitis por reflujo, síndrome de Zollinger – Ellison. Coadyuvante en la erradicación del Helicobacter pylori en úlcera péptica. Reflujo gastroesofágico sintomático. Dispepsia ácida. Úlceras o erosiones gástricas y duodenales relacionadas con AINEs.
VÍA DE ADMINISTRACIÓN: Oral
FORMA FARMACÉUTICA: Cápsula
DOSIFICACIÓN: En pacientes con úlcera péptica el régimen estándar de dosificación es de 20 mg durante 4 semanas. Cuando no se logre la respuesta en este plazo se sugiere continuar la terapia otras 4 semanas. Se han administrado dosis hasta de 40 mg diarios durante 8 semanas para conseguir cicatrización. En pacientes con úlcera duodenal el régimen estándar de dosificación, es de 20 mg durante 4 semanas. En pacientes con esofagitis por reflujo el régimen estándar de dosificación es de 20 mg durante 4 semanas. Cuando no se logre la respuesta en este plazo se sugiere continuar la terapia durante otras 4 semanas. En pacientes con sindrome de Zollinger-Ellison la dosis inicial recomendada es de 60 mg en una sola toma diaria. De acuerdo a la respuesta clínica del paciente se determinará la dosis que de acuerdo a la severidad puede llegar hasta 120 mg diarios. Cuando la dosis utilizada excede de 80 mg se recomienda dividir en 2 tomas diarias.
CONTRAINDICACIONES: Hipersensibilidad conocida al omeprazol, benzimidazoles sustituidos o a cualquier otro componente de la formulación. PRECAUCIONES Y ADVERTENCIAS: Si surge algún síntoma alarmante (por ejemplo, pérdida de peso considerable e involuntaria, vómito recurrente, disfagia, hematemesis o melena) y si se confirma o se sospecha de una úlcera gástrica, debe descartarse la presencia de una enfermedad maligna ya que el tratamiento puede aliviar los síntomas y retrasar su diagnóstico. Los pacientes tratados con omeprazol durante periodos prolongados de tiempo tienen el riesgo de generar niveles bajos de magnesio sérico (hipomagnesemia) la cual puede manifestarse con alteraciones de la frecuencia cardiaca (palpitaciones rápidas) u otros síntomas como espasmos musculares temblores o convulsiones: En los niños, las tasas anormales del corazón pueden causar fatiga, malestar estomacal, mareos y aturdimiento. Evítese el consumo concomitante con medicamentos como furosemida, ácido etacrínico, clorotiazida, hidroclorotiazida, indapamina y metolazona. El omeprazol puede reducir la actividad farmacológica del clopidogrel, debiéndose ajustar las dosis. Pregúntele a su médico o farmacéutico sobre el uso de este medicamento si usted está tomando warfarina, clopidogrel o cilostazol (anticoagulantes) o antirretrovirales recetados (medicamentos para la infección por el VIH). Los inhibidores de la bomba de protones (IBP) se asocian a casos muy infrecuentes de una reacción conocida como lupus eritematoso cutáneo subagudo (LECS), esta reacción se caracteriza por lesiones eritematosas en zonas expuestas al sol acompañadas de artralgias. En caso de presentarse, se debe solicitar atención médica y considerar la interrupción del tratamiento con PRAZED®. La aparición de LECS con el tratamiento de IBP previo, puede aumentar el riesgo de LECS con otros IBP.
INTERACCIONES: Principios activos con una absorción dependiente del pH: Nelfinavir, atazanavir: Las concentraciones plasmáticas de nelfinavir y atazanavir disminuyen cuando se administran conjuntamente con omeprazol. No se recomienda la administración concomitante de omeprazol y atazanavir El aumento de la dosis de atazanavir a 400 mg no compensó el efecto del omeprazol en la exposición a atazanavir. Digoxina: El tratamiento concomitante de omeprazol (20 mg al día) y digoxina en sujetos sanos aumentó la biodisponibilidad de la digoxina en un 10%. Rara vez se han comunicado casos de toxicidad por digoxina. Se recomienda precaución cuando se administre omeprazol en dosis altas en ancianos, por tener la digoxina un estrecho margen terapéutico. Clopidogrel: En un estudio clínico cruzado, se administró clopidogrel (dosis de carga de 300 mg seguidos de 75 mg/día) solo y con omeprazol (80 mg al mismo tiempo que el clopidogrel) durante 5 días. La exposición al metabolito activo del clopidogrel disminuyó en un 46% (día 1) y 42% (día 5) cuando el clopidogrel y el omeprazol se administraron juntos. La inhibición media de agregación plaquetaria (IAP) disminuyó en un 47% (24 horas) y 30% (Día 5) cuando el clopidogrel y el omeprazol se administraron juntos. En otro estudio, se demostró que administrar el clopidogrel y el omeprazol en distintos momentos no impedía su interacción, ya que es probable que esté impulsada por el efecto inhibitorio del omeprazol sobre CYP2C19. Otros principios activos: La absorción de posaconazol, erlotinib, ketoconazol e itraconazol se reduce de forma significativa, por lo que pueden perder eficacia clínica. Se recomienda evitar la administración concomitante con posaconazol y erlotinib. Principios activos metabolizados por CYP2C19: El omeprazol es un inhibidor moderado de CYP2C19, su principal enzima metabolizadora. Por lo tanto, puede disminuir el metabolismo de principios activos concomitantes también metabolizados por CYP2C19 y aumentar la exposición sistémica como el caso de la D-warfarina y otros antagonistas de la vitamina K, como cilostazol, diazepam y fenitoína. Fenitoína: Como es de estrecho margen terapéutico, se recomienda vigilar niveles plasmáticos durante las dos primeras semanas después de iniciar el tratamiento con omeprazol y, si se realiza un ajuste de la dosis de fenitoína, debe realizarse una monitorización y un nuevo ajuste de la dosis al finalizar el tratamiento con omeprazol. Mecanismo desconocido: La administración concomitante de omeprazol y saquinavir/ritonavir aumentó la concentración plasmática de saquinavir en casi un 70% y se asoció a una buena tolerancia en los pacientes infectados por el VIH. Tacrolimus: Se ha comunicado que la administración concomitante de omeprazol puede aumentar la concentración sérica de tacrolimús. Se recomienda monitorización de las concentraciones de tacrolimus así como de la función renal (aclaramiento de creatinina) y ajustar la dosis de tacrolimús en caso necesario. Efectos de otros principios activos sobre la farmacocinética del omeprazol Inhibidores de CYP2C19 y/o CYP3A4. Puesto que el omeprazol es metabolizado por CYP2C19 y CYP3A4, medicamentos como claritromicina y voriconazol que inhiben CYP2C19 o CYP3A4 pueden conducir a un aumento de las concentraciones séricas del omeprazol. El tratamiento concomitante con voriconazol ha tenido como resultado un aumento de la exposición al omeprazol de más del doble. Como las dosis altas de omeprazol se toleran bien, generalmente no es necesario un ajuste de dosis. Sin embargo, en pacientes con insuficiencia hepática grave y si está indicado un tratamiento de larga duración, debe considerarse un ajuste de la dosis. Inductores de CYP2C19 y/o CYP3A4: Los principios activos como la rifampicina y la hierba de San Juan que inducen CYP2C19 o CYP3A4, pueden disminuir las concentraciones séricas de omeprazol.
EFECTOS SECUNDARIOS: Dermatológicos: Raramente ocurrirán erupción y/o prurito. En casos aislados se han descrito fotosensibilidad, eritema multiforme, alopecia. Musculoesqueléticas: Casos aislados de artralgia, debilidad muscular y mialgias. SNC: Cefalea, parestesias, somnolencia, insomnio, vértigo. En casos aislados se han descrito confusión mental reversible, agitación, depresión, y alucinaciones, principalmente en pacientes en estado grave. Gastrointestinales: Diarrea, constipación, dolor abdominal, náusea, vómito y flatulencia. Se han reportado casos aislados de estomatitis y candidiasis gastrointestinal. Hepáticos: Se han reportado raramente elevación transitoria de enzimas hepáticas. En casos aislados puede ocurrir encefalopatía hepática en pacientes con insuficiencia hepática grave pre-existente por hepatitis con o sin ictericia. Endocrinos: Se han reportado casos aislados de ginecomastia. Hematológicos: Casos aislados de leucopenia, trombocitopenia, agranulocitosis y pancitopenia. Otros: Raramente pueden ocurrir reacciones de hipersensibilidad, nefritis interstiticial, trastornos visuales irreversibles en pacientes gravemente enfermos, especialmente cuando han recibido dosis elevadas, no se estableció una relación causal.
PROPIEDADES FARMACODINÁMICAS: El omeprazol reduce la secreción de ácido a nivel gástrico mediante un mecanismo de acción altamente selectivo, consistente en la inhibición de la H+K+ATPasa (bomba de ácido de protones) en la célula parietal gástrica en forma dosis-dependiente, actuando rápidamente y produciendo un control reversible de la secreción ácida del estomago, controlando la secreción basal de ácido así como la inducida por estímulos. En los canalículos secretores ácidos de la celula parietal adyacentes a la bomba de ácido, la molécula de omeprazol por ser una base débil, gana hidrogeniones y se transforma en un inhibidor activo. El omeprazol es una base débil que se concentra en el medio ácido de los canalículos secretores de la célula parietal gástrica donde es convertido por el ácido en el derivado activo sulfonamida. Tal molécula al interactúar con grupos sulfhidrilo de la bomba de protones, la inhibe activamente bloqueando irreversiblemente la secreción de hidrogeniones al lumen gástrico. El omeprazol inhibe la bomba de protones solamente con valores de pH inferiores a 4, siendo inactivo en condiciones de pH superior a esta cifra.
CONDICIÓN DE VENTA: Venta con fórmula médica
PRESENTACIÓN: Caja por 21 cápsulas conteniendo 20 mg de omeprazol en microgránulos con cubierta entérica. Reg. San. INVIMA 2019M-0013109-R1
Lanzor®
REGISTRO SANITARIO:
INVIMA 2014M-0014820
PRINCIPIO ACTIVO:
Lansoprazol
UNIDAD DE VENTA:
Cápsulas con Microgránulos
COMPOSICIÓN: Cada cápsula contiene 30 mg de lansoprazol en microgránulos; excipientes c.s.
INDICACIONES: Medicamento alternativo en el manejo de la úlcera péptica, esofagitis por reflujo y síndrome de Zollinger-Ellison. Puede ser usado en niños mayores de 1(un) año de edad.
VÍA DE ADMINISTRACIÓN: Oral
DOSIFICACIÓN: La dosis recomendada para el tratamiento de la úlcera duodenal y de la esofagitis por reflujo es de 30 mg diarios, en una única toma lejos de los alimentos. La duración media estimada del tratamiento es de 2 a 4 semanas para el tratamiento de la úlcera duodenal y de 4 a 8 semanas para la úlcera gástrica o esofagitis por reflujo. En pacientes con síndrome de Zollinger-Ellison deberá individualizarse la dosis efectiva para mantener la secreción ácida entre 0 y 10mmol/h pero puede llegarse a dosis de 60 a 120 mg diarios. En la enfermedad ulcerosa gastroduodenal que no cicatriza en un período de 4 a 8 semanas, se puede intentar aumentar la dosis a 60 mg/día y controlar vía endoscópica la lesión.
CONTRAINDICACIONES Y ADVERTENCIAS: Lansoprazol está contraindicado en pacientes con conocida hipersensibilidad a alguno de los componentes de la formulación, embarazo, lactancia, menores de un año, úlcera gástrica de origen neoplásico o sin diagnóstico definido. Lansoprazol no debe ser coadministrado con atazanavir ya que el uso concomitante puede producir una reducción significante en la exposición del atazanavir. PRECAUCIONES Y ADVERTENCIAS: Malignidad gástrica: La respuesta sintomática al tratamiento con lansoprazol no excluye la presencia de malignidad gástrica. Fracturas óseas: Varios estudios observacionales publicados sugieren que los inhibidores de la bomba de protones (IBP) pueden estar asociados con un mayor riesgo de fracturas osteoporóticas de cadera, muñeca o columna vertebral. El riesgo de fractura fue mayor en los pacientes que recibieron altas dosis, definidas como dosis diarias múltiples y tratamiento de largo plazo con IBP (un año o más). Los pacientes deben utilizar la dosis más baja y la menor duración de la terapia de IBP, adecuada para la enfermedad que padecen. Pacientes con riesgo de fracturas osteoporóticas deben ser manejados de acuerdo a las pautas de tratamiento establecidas. Hipomagnesemia: La hipomagnesemia, sintomática y asintomática, ha sido raramente reportada en pacientes tratados con IBP al menos durante tres meses, en la mayoría de los casos después de un año de terapia. Los eventos adversos graves incluyen tétano, arritmias y convulsiones. En la mayoría de los pacientes, el tratamiento para la hipomagnesemia requirió reemplazo de magnesio e interrupción de la terapia con IBP. Para los pacientes que se espera estén en tratamiento prolongado o que toman IBP con medicamentos como la digoxina o medicamentos que pueden causar hipomagnesemia (por ejemplo, diuréticos de asa como furosemida, torasemida, bumetanida y ácido etacrínico o tiazídicos como clorotiazida, hidroclorotiazida, indapamida y metolazona), los profesionales de la salud deben considerar el monitoreo de los niveles de magnesio antes de iniciar el tratamiento con IBP y controlar los niveles periódicamente. Se requiere de atención inmediata si se experimentan síntomas como frecuencia o ritmo cardíaco anormal, o síntomas tales como palpitaciones rápidas, palpitaciones, espasmos musculares, temblores o convulsiones mientras toma un medicamento IBP. En los niños, las tasas anormales del corazón pueden causar fatiga, malestar estomacal, mareos y aturdimiento. Los inhibidores de la bomba de protones (IBP) se asocian a casos muy infrecuentes de una reacción conocida como lupus eritematoso cutáneo subagudo (LECS), esta reacción se caracteriza por lesiones eritematosas en zonas expuestas al sol acompañadas de artralgias. En caso de presentarse, se debe solicitar atención médica y considerar la interrupción del tratamiento con el LANZOR 30 mg. La aparición de LECS con el tratamiento de IBP previo, puede aumentar el riesgo de LECS con otros IBP.
EFECTOS SECUNDARIOS: El lansoprazol es bien tolerado. Raramente se han comunicado diarreas, constipación, náuseas y cefaleas. En algún paciente ocasionalmente ha aparecido rash cutáneo. Generalmente estos síntomas son leves y transitorios, sin que se haya podido establecer una relación de causalidad con el tratamiento. Hallazgos de laboratorio: Raras elevaciones de eosinófilos, triglicéridos, enzimas hepáticos y de la potasemia fueron observadas, sin encontrarse una correlación con la dosis ni con la duración del tratamiento. El lansoprazol puede producir una elevación moderada de la gastrinemia, la cual vuelve a la normalidad al mes siguiente de finalizar el tratamiento. Además, después de finalizar el tratamiento con lansoprazol los resultados obtenidos de las biopsias gástricas practicadas no muestran elementos que sugieran un tumor carcinoide o una proliferación celular. No hay datos disponibles sobre intoxicación en el hombre con lansoprazol por lo que únicamente se puede recomendar tratamiento sintomático.
INTERACCIONES: El lansoprazol es metabolizado por el sistema del citocromo hepático P450, concretamente mediante las isoenzimas CYP3A y CYP2C19 y puede dar lugar a una ligera inducción del sistema enzimático monooxigenasa del citocromo P450. No se han observado interacciones del lansoprazol con antipirina, claritromicina, diazepam, ibuprofeno, indometacina, fenitoína, prednisona, propranolol, terfenadina o warfarina en individuos normales. El uso concomitante de la teofilina (un sustrato para CYP3A y CYP2C19) y del lansoprazol indujo un pequeño aumento (10%) en el aclaramiento de la teofilina. Puede requerirse un pequeño ajuste en las dosis de teofilina si se añade o discontinua un tratamiento con lansoprazol. La administración simultánea de antiácidos (conteniendo hidróxido de aluminio y magnesio) con lansoprazol modifica los parámetros farmacocinéticos y la biodisponibilidad de éste, por lo que se aconseja su administración con posterioridad al antiácido (1 hora). El sucralfato ha demostrado reducir la absorción y biodisponibilidad del lansoprazol en un 17%. El lansoprazol deberá ser administrado no menos de 30 minutos antes del sucralfato se debe usar ambos fármacos concomitantemente. El lansoprazol posee efectos duraderos sobre la secreción gástrica de ácido. Para aquellos fármacos cuya biodisponibilidad sea afectada por el pH gástrico, la administración concomitante del lansoprazol puede ejercer un efecto significativo sobre su absorción. Entre los fármacos que pueden ser afectados de esta manera por el lansoprazol se encuentran la ampicilina, las sales de hierro, el itraconazol, y el ketoconazol. El lansoprazol ha mostrado no producir una interacción clínicamente significativa con la amoxicilina. Los inhibidores de la bomba ácida gástrica pueden aumentar la biodisponibilidad de la digoxina. Sin embargo la magnitud de este efecto es pequeña. La potencial interacción entre el lansoprazol y la digoxina no ha sido estudiada específicamente. Los pacientes con niveles séricos de digoxina en el límite superior o inferior deberán ser monitorizados de cerca si se administra un inhibidor de la bomba de protones con la digoxina. Se ha estudiado el tratamiento crónico conjunto con lansoprazol y diazepam. La semivida de eliminación, el aclaramiento y el volumen de distribución del diazepam no estuvieron afectados por el uso concomitante el lansoprazol. Tampoco se han encontrado interacciones significativas en un estudio controlado por placebo de dosis múltiples de lansoprazol con anticonceptivos orales conteniendo etinilestradiol y levonorgestrel. No se modificó la biodisponibilidad de los anticonceptivos.
PROPIEDADES FARMACODINÁMICAS: Es un derivado benzimidazólico de segunda generación, que se diferencia de su antecesor, el omeprazol, por presentar un grupo trifluoroetoxi en su estructura química base, lo que le otorga una mayor biodisponibilidad (30%) y potencia farmacológica. Por su mayor liposolubilidad atraviesa con más facilidad la membrana de la célula parietal gástrica responsable de la producción de ácido clorhídrico en la cual se transforma en los metabolitos activos disulfuro y sulfonamida, que al unirse con los grupos sulfhidrilos (SH) de la bomba de protones H+/K+-ATPasa, inhiben la secreción acido péptica. En estudios recientes se señala que el lansoprazol ejerce un efecto bactericida sobre el Helicobacter pylori, microorganismo gramnegativo flagelado al que se atribuye un papel en la producción de gastritis crónica y en la recidiva de la enfermedad ulcerosa gastroduodenal. El efecto inhibitorio sobre el H. pylori se logra con concentraciones inhibitorias mínimas (CIM90) muy bajas (6,25 mg/mL) si se las compara con las de otros fármacos (subcitrato de bismuto, ranitidina u omeprazol). En estudios farmacodinámicos experimentales, en roedores (rata) y no roedores (perro), el lansoprazol mostró una potencia 10 veces mayor que el omeprazol en la prevención de la úlcera experimental (por ácido acético, aspirina o ligadura pilórica). Con dosis única de 30 mg por vía oral se logra una marcada inhibición de la secreción acidopéptica con mantenimiento del pH intragástrico en valores mayores que 3. El lansoprazol es un fármaco ácido-lábil, por lo tanto, para su empleo por vía oral se usan cápsulas con microgránulos gastrorresistentes, tal como ocurre con el omeprazol.
CONDICIÓN DE VENTA: Venta con fórmula médica
PRESENTACIÓN: Caja por 28 cápsulas Reg. San. INVIMA 2014M-0014820.

Expectos®
REGISTRO SANITARIO:
INVIMA PFM2020-0002726
PRINCIPIO ACTIVO:
Hedera helix 1%
UNIDAD DE VENTA:
Jarabe
COMPOSICIÓN: Cada 100 mL de jarabe contienen 1,0 g de extracto seco (5 – 7,5: 1) de hojas de hiedra (Hedera helix L.) con un contenido de no menos de 90 mg de Hederacósido C, excipientes c.s. Cada 5 mL de jarabe contienen 50 mg de extracto seco (5 – 7,5: 1) de hojas de hiedra (Hedera helix L.), excipientes c.s.
USOS TERAPÉUTICOS: Expectorante. Coadyuvante en el tratamiento de la tos.
VÍA DE ADMINISTRACIÓN: Oral.
FORMA FARMACÉUTICA: Jarabe.
DOSIFICACIÓN: Niños de 2 a 5 años: 1,7 mL, dos (2) veces al día. Niños de 6 a 12 años: 3,5 mL, dos (2) veces al día. Adolescentes y adultos: 3,5 – 5,2 mL, dos (2) veces al día. Las cajas van con dosificador que permite la dosificación antes descrita. No exceder de las dosis recomendadas.
CONTRAINDICACIONES: Hipersensibilidad a algunos de sus componentes. Embarazo y lactancia. En caso de intolerancia a la fructosa, el tratamiento sólo debe realizarse después de consultar al médico.
EFECTOS SECUNDARIOS: Aunque no son comunes, en individuos sensibles a alguno de sus componentes, podrían llegar a presentarse malestares gastrointestinales (náuseas, vómitos, diarrea) o reacciones alérgicas (urticaria, erupción cutánea, disnea).
PROPIEDADES FARMACODINÁMICAS: El efecto mucolítico y secretolítico se debe esencialmente a la naturaleza de las saponinas, las cuales presentan propiedades fluidificantes del moco, facilitando, también, el aclaramiento ciliar. En el extracto de Hedera helix se identifican tres saponinas principales: La hederagenina, la alfa-hederina y el hederacósido C.
Hederagenina: La hederagenina no posee cadena de azúcares. Los estudios realizados mostraron que esta molécula no actúa sobre la fisiología pulmonar. La alfa-hederina es el componente activo más importante del extracto de Hedera helix, según se pudo observar en las experiencias realizadas con esta molécula. Hederacósido C: El hederacósido C carece de actividad directa sobre la fisiología bronquial, sin embargo, es muy importante porque actúa como un profármaco que produce un clivaje en la cadena de azúcar de la molécula por acción de esterasas de la sangre, lo que da origen a alfa-hederina. El efecto mucolítico es ejercido a través del efecto sobre la tensión superficial del moco, mientras que el efecto secretolítico es iniciado por vía parasimpática por estimulación refleja sobre elementos celulares del bronquio y glándulas bronquiales. Consecuentemente, se produce un aumento del componente líquido, el cual conduce a la reducción de la viscosidad del moco, facilitando así su remoción. El efecto broncoespasmolítico es ejercido por una inhibición de la internalización de los receptores ß2 adrenérgicos presentes en las células de músculo liso bronquial los cuales al permanecer más tiempo en la superficie celular son estimulados y llevan a una dilatación bronquial. Se ha demostrado que la mayoría de las saponinas presentan efectos broncoespasmolíticos, habiéndose cuantificado a través de cromatografía de columna. De ahí ha surgido que el efecto broncoespasmolítico de los diferentes compuestos aislados de la Hedera helix son aditivos. El efecto broncodilatador del extracto de Hedera helix tres horas después de su ingestión es comparable con el de la inhalación de un beta2 simpaticomimético. La propiedad antitusiva del extracto resulta como consecuencia natural de las acciones mucolítica y espasmolítica, eliminando secundariamente la tos sin causar deterioro en el control central de la respiración.
CONDICIÓN DE VENTA: Venta sin fórmula facultativa
PRESENTACIÓN: Frasco por 120 mL. Reg. San. INVIMA PFM2020-0002726.

Lordinex® D
REGISTRO SANITARIO:
INVIMA. 2018M-0012862-R1
PRINCIPIO ACTIVO:
loratadina
UNIDAD DE VENTA:
Cápsulas
Cápsulas
Antihistamínico antiH1,
descongestionante
COMPOSICIÓN: Cada CÁPSULA de LORDINEX® D contiene loratadina 5 mg, fenilefrina clorhidrato en microgránulos 20 mg, excipientes csp
INDICACIONES: Antihistamínico antiH1, descongestionante.
VÍA DE ADMINISTRACIÓN: Oral
DOSIFICACIÓN: 1 cápsula cada 12 horas
CONTRAINDICACIONES: Historia de hipersensibilidad a los componentes de la formulación. Pacientes con enfermedad renal terminal con una aclaración de creatinina inferior a 10 mL/ min. El producto puede presentar interacción con IMAOS, por lo tanto debe evitarse su uso concomitante. Niños menores de 12 años. Debe ser evitado en pacientes con hipertensión, arterioesclerosis avanzada, aneurisma, hipotensión ortostática y diabetes mellitus dependiente de insulina. Su potencial de toxicidad y abuso es bajo. Mantener fuera del alcance de los niños. La utilización de fenilefrina durante el período final del embarazo o durante el parto puede ocasionar anoxia y bradicardia fetal por aumento de la contractilidad uterina y disminución del flujo sanguíneo uterino. Puede ocurrir bradicardia refleja, efectos sobre el SNC, no se recomienda en pacientes hipertensos, con enfermedad cardiovascular con o sin arritmia, enfermedad cerebrovascular o síndrome orgánico cerebral, pacientes con hipertiroidismo y con hiperplasia prostática benigna.
INTERACCIONES: Loratadina: La loratadina se ha coadministrado con eritromicina, cimetidina y ketoconazol, y aunque se reporta aumento de área bajo la curva de loratadina (ABC de 32 a 91 mcg/L x h coadministrada con ketoconazol), esta interacción carece de significado clínico, por ausencia de efectos en el electrocardiograma, pruebas de laboratorio, signos vitales o efectos adversos. No hubo modificación en el intervalo QT cardíaco. El area bajo la curva de eritromicina disminuyó en 15% al comparar con la curva de eritromicina sola, desconociéndose la relevancia clínica de este hallazgo. No se reportaron sedación ni síncope. Debe suspenderse aproximadamente 48 horas antes de efectuar cualquier tipo de prueba cutánea, ya que los antihistamínicos pueden impedir o disminuir las reacciones que, de otro modo, serían positivas a los indicadores de reactividad dérmica. Fenilefrina: El uso simultáneo con anestésicos orgánicos por inhalación (cloroformo, ciclopropano, enflurano, halotano, isoflurano) puede aumentar el riesgo de arritmias ventriculares severas. Se reduce el efecto de drogas antihipertensivas y de diuréticos empleados como antihipertensivos. El uso junto con aminoglucósidos digitálicos o levodopa también aumenta el riesgo de arritmias cardíacas. La oxitocina, dihidroergotamina y ergometrina pueden ocasionar un aumento de la vasoconstricción. Los antidepresivos tricíclicos e inhibidores de la MAO pueden potenciar los efectos cardiovasculares de la fenilefrina. El uso simultáneo de hormonas tiroideas puede aumentar los efectos de la hormona o de la fenilefrina. Puede reducir los efectos antianginosos de los nitratos.
EFECTOS SECUNDARIOS: Loratadina: No hubo diferencias estadísticamente significativas en la ocurrencia de efectos anticolinérgicos, como fatiga, sedación, sequedad oral, cefalea al comparar frente a placebo, ni en la tasa de abandonos en el grupo de tratamiento activo frente al de placebo. Fenilefrina: Signos de sobredosis: Taquicardia, palpitaciones, cefalea, hormigueo en manos y pies, vómitos. Pueden aparecer como efectos secundarios: Mareos, nerviosismo, temblor, disnea, debilidad no habitual.
PROPIEDADES FARMACODINÁMICAS: Loratadina: Es un antihistamínico tricíclico de larga acción con actividad antagonista altamente selectiva a nivel de los receptores H1 periféricos de histamina. La actividad antialérgica se ha estudiado empleando basófilos de humanos de sujetos alérgicos y no alérgicos, encontrándose inhibición de liberación de histamina inducida por IgE. Adicionalmente, estabiliza la membrana celular del mastocito. No se han observado efectos anticolinérgicos. Fenilefrina: Amina simpaticomimética de acción directa, aunque también actúa en forma indirecta mediante la liberación de noradrenalina de los lugares de almacenamiento. Como vasopresor actúa sobre los receptores alfa-adrenérgicos para producir vasoconstricción. En dosis terapéuticas produce muy poca estimulación del SNC. Es un agonista de los receptores alfa y la mayoría de su acción simpaticomimética es debida principalmente a estimulación directa de los receptores alfa, con poca liberación de noradrenalina. Es un agonista de los receptores alfa y la mayoría de su acción simpaticomimética es debida principalmente a estimulación directa de los receptores alfa, con poca liberación de noradrenalina. Dada entonces su poca liberación de noradrenalina, a dosis terapéuticas no causa estimulación significativa del sistema nervioso central y es segura a nivel cardiovascular, siendo el vasoconstrictor más seguro en pacientes con hipertensión arterial.
CONDICIÓN DE VENTA: Venta bajo fórmula médica
PRESENTACIÓN: Caja por 10 cápsulas. Reg. San. INVIMA. 2018M-0012862-R1.
Cronofen® Tabletas
REGISTRO SANITARIO:
INVIMA 2020M-0013210-R1
PRINCIPIO ACTIVO:
Acetaminofén
UNIDAD DE VENTA:
Tabletas
Otros analgésicos y
antipiréticos
(Acetaminofén)
COMPOSICIÓN: Cada tableta contiene Acetaminofén 500 mg y excipientes c.s.
INDICACIONES: Analgésico, Antipirético
VÍA DE ADMINISTRACIÓN: Oral
Usos: Para el alivio de dolores: Dolor de cabeza, migraña, dismenorrea, dolor de garganta, dolor muscular, dolor postraumático, dolor de oídos, dolor dental, dolor neurálgico, dolor posquirúrgico, dolor y fiebre asociados al resfriado común, dolor y fiebre después de la vacunación, dolor por osteoartritis, otros estados febriles o dolorosos.
FORMA FARMACÉUTICA: Tableta
DOSIFICACIÓN: Adultos: Tomar 1 a 2 tabletas 3 veces al día. Niños de 7 a 12 años: Tomar 1/2 a 1 tableta 3 veces al día.
CONTRAINDICACIONES: Hipersensibilidad al acetaminofén o a sus excipientes. PRECAUCIONES Y ADVERTENCIAS: No debe administrarse junto con otros medicamentos que contengan acetaminofén soló o en combinación con otros medicamentos, ya que puede conducir a una sobredosis. Los riesgos de sobredosis son mayores en aquellos con enfermedad hepática alcohólica no cirrótica. Se recomienda precaución en la administración de acetaminofén a pacientes con insuficiencia renal o hepática grave, con deficiencia conocida de glucosa-6-fosfato-deshidrogenasa y/o síndrome de Gilbert. En pacientes con diagnóstico previo de enfermedad hepática o renal, debe existir una evaluación médica antes de tomar este medicamento que contiene acetaminofén. Si los síntomas persisten o empeoran, consulte a su médico. Consulte con su médico de inmediato si usted supera la posología indicada, incluso si se siente bien, debido a que el consumo excesivo de acetaminofén o el de 3 o más bebidas alcohólicas por día, puede causar daño grave al hígado a largo plazo. Acetaminofén puede causar reacciones cutáneas graves, si se produce una reacción, como enrojecimiento de la piel, ampollas o erupción, deben suspender y buscar asistencia médica de inmediato. Niños menores de 12 años no debe ser indicado. El esquema posológico con acetaminofén no deberá superar los 3 g por día, repartidas en concentraciones que no proporcionen más de 500 mg. Por toma. Para los niños mayores de 12 años la dosis no debe superar los 40 mg/kg por día, repartido en concentraciones que no proporcionen más de 10 mg/kg por toma.
INTERACCIONES: El riesgo de toxicidad con paracetamol (acetaminofén) se puede incrementar al suministrar concomitantemente con otros fármacos potencialmente hepatotóxicos o que induzcan las enzimas microsomales hepáticas. La absorción del paracetamol se puede acelerar con la metoclopramida. La administración de probenecid puede alterar las concentraciones plasmáticas del paracetamol. La colestiramina disminuye la absorción del paracetamol. Se ha descrito hepatotoxicidad en pacientes que reciben isoniazida. Se puede incrementar el riesgo de sangrado en pacientes que reciben anticoagulantes. Se han descrito casos de hepatotoxicidad o neutropenia en pacientes que recibían zidovudina, aunque estos hallazgos no han sido consistentes. El paracetamol aumenta la eliminación urinaria de la lamotrigina.
EFECTOS SECUNDARIOS: Suspenda el medicamento y consulte inmediatamente al médico si: 1, se presentan reacciones alérgicas como erupciones cutáneas o prurito (picazón), algunas veces con problemas de respiración o inflamación de labios, lengua, garganta o cara. 2. Se presenta salpullido o peladuras en la piel o úlceras en la boca. 3. Usted ha sufrido previamente de problemas en la respiración cuando ha ingerido ácido acetil salicílico u otros AINE y se presenta una reacción similar con este producto. 4. Se presenta sangrado inesperado al cepillarse 5. Estas reacciones son raras. Toxicidad de acetaminofén: Una dosis única mayor de 120 mg/kg se ha considerada como la necesaria para causar intoxicación por este analgésico. Este dato se obtuvo originalmente de adultos que ingirieron sobredosis de acetaminofén con fines suicidas El comportamiento en niños es diferente: Dosis de 50 a 60 mg/kg, administradas en formas repetidas o bien, dosis terapéuticas en niños con enfermedad hepática, son capaces de producir intoxicaciones graves.43En estas condiciones la producción del metabolito se incrementa lo que causa depleción del glutatión; cuando ésta es menor a 50%, la n -acetilimidoquina penetra al hepatocito donde se une en forma covalente con las macromoléculas dando como resultado final necrosis celular. Se piensa que por este mismo mecanismo de depleción de glutatión, se produce daño simultáneo en el riñón y el miocardio. Otros factores que pueden influir en la toxicidad del acetaminofén son la inmadurez funcional hepática de los neonatos y lactantes menores, la administración concomitante de fármacos inductores del sistema microsomal hepático como el fenobarbital y el etanol (empleado en los jarabes y elíxires de uso pediátrico) o bien fármacos hepatotóxicos per se como el ácido valproico o la eritromicina. Finalmente, dada la gravedad y alta mortalidad con que puede evolucionar esta intoxicación, se han establecido criterios para considerar el trasplante hepático: 47pH menor de 7.3, tiempo de protombina mayor de 100 segundos, creatinina sérica mayor de 300 µmol/l, coma grado 3 o 4. Manejo de sobredosis de acetaminofén: El efecto adverso más grave descripto con la sobredosis aguda de paracetamol es una necrosis hepática, dosis-dependiente, potencialmente fatal. La necrosis hepática (y la tubular renal) son el resultado de un desequilibrio entre la producción del metabolito altamente reactivo y la disponibilidad de glutatión. Con disponibilidad normal de glutatión, la dosis mortal de paracetamol es de 10g aproximadamente; pero hay varias causas que pueden disminuir estas dosis (tratamiento concomitante con doxorrubicina o el alcoholismo crónico). El tratamiento debe comenzarse con N-acetilcisteína por vía intravenosa sin esperar a que aparezcan los síntomas, pues la necrosis es irreversible.
PROPIEDADES FARMACODINÁMICAS: Actúa por inhibición de la síntesis de prostaglandinas a nivel del sistema nervioso central, y a diferencia de los AINE, tiene un bajo efecto inhibitorio de la síntesis periférica de éstas.
CONDICIÓN DE VENTA: Venta libre
PRESENTACIÓN: CRONOFEN® tabletas caja por 100 y por 50 unidades en blister. Reg. San. No. INVIMA 2020M-0013210-R1.
Cronofen® Forte
REGISTRO SANITARIO:
INVIMA 2026M-002574
PRINCIPIO ACTIVO:
Acetaminofén 500mg y Cafeína 65mg
UNIDAD DE VENTA:
Tabletas
Analgésico – antipirético. Contraindicaciones hipersensibilidad conocida al acetaminofén, cafeína o a cualquiera de los excipientes de la formulación. Insuficiencia hepática severa o enfermedad hepática activa severa. Alteraciones cardiovasculares graves. Hipertensión grave no controlada. Acetaminofén: al igual que con todos los analgésicos deben evitarse tratamientos muy prolongados o dosis mayores a las recomendadas. El acetaminofén deberá darse con cuidado a pacientes que tienen deterioro de la función renal o hepática (en este último caso, el uso ocasional es aceptable, pero la administración prolongada de dosis elevadas puede aumentar el riesgo de aparición de efectos adversos). Puede ocurrir hepatotoxicidad con el acetaminofén aún en dosis terapéuticas, después de períodos cortos de tratamiento y en pacientes con enfermedad hepática pre-existente. Deberá administrarse con precaución a los pacientes que tienen dependencia de alcohol, malnutrición crónica, anoréxicos, con bajo índice de masa corporal o deshidratación. Debe tenerse precaución en los pacientes que tengan una hipersensibilidad subyacente al ácido acetilsalicílico y/o medicamentos antiinflamatorios no esteroideos (aines). Se recomienda precaución en pacientes asmáticos sensibles al ácido acetilsalicílico, debido a que se han descrito reacciones broncoespásticas con acetaminofén (reacción cruzada) en estos pacientes, aunque sólo se han manifestado en una minoría de dichos pacientes, puede provocar reacciones graves en algunos casos, especialmente cuando se administra en dosis altas. Reacciones cutáneas severas: se han reportado reacciones cutáneas que pueden amenazar la vida como el síndrome de stevens- johnson (ssj) y la necrólisis epidérmica tóxica (net) con el uso de acetaminofén. Se debe informar a los pacientes sobre los signos y síntomas y monitorear de cerca las reacciones cutáneas. Si se presentan signos y síntomas de ssj y net (por ejemplo, rash o erupción cutánea frecuentemente con ampollas o lesiones en mucosas) los pacientes deben suspender inmediatamente el tratamiento con acetaminofén y consultar al médico. Acetaminofén se debe administrar con precaución, evitando tratamientos prolongados en pacientes con anemia, afecciones cardíacas o pulmonares. En pacientes con estados de deficiencia de glutatión como sepsis, el uso de acetaminofén puede incrementar el riesgo de acidosis metabólica. Se debe limitar la automedicación con acetaminofén cuando se está en tratamiento con anticonvulsivantes debido a que con el uso concomitante de ambos se potencia la hepatotoxicidad y se disminuye la biodisponibilidad del acetaminofén, especialmente en tratamientos con dosis altas de acetaminofén. Debe advertirse al paciente que evite el uso simultáneo de este medicamento con otros que contengan acetaminofén, como por ejemplo medicamentos antigripales. En caso de administrarse otro medicamento que contenga acetaminofén no se deberá exceder la dosis máxima de acetaminofén de 3g al día teniendo en cuenta el contenido del mismo de todos los medicamentos que utiliza el paciente. El uso simultáneo de más de un medicamento que contenga acetaminofén, puede dar lugar a cuadros de intoxicación. Los cuadros tóxicos asociados a acetaminofén se pueden producir tanto por la ingesta de una sobredosis única o por varias tomas con dosis excesivas de acetaminofén. Si hay alguna sospecha, aunque modesta, de una sobredosis de acetaminofén, un médico debe ser contactado inmediatamente. Cafeína: se debe administrar bajo control médico en aquellos pacientes con arritmias cardíacas, hiperfunción tiroidea y pacientes con síndromes ansiosos. En pacientes que hayan sufrido un infarto de miocardio, se recomienda no administrar cafeína hasta que hayan trascurrido varias semanas desde el accidente. La cafeína puede elevar los niveles de glucosa en sangre por lo que deberá tenerse en cuenta en pacientes diabéticos. Los pacientes sensibles a otras xantinas (aminofilina, teofilina, entre otros) también pueden ser sensibles a la cafeína por lo que no deberían tomar este medicamento. Se debe tener precaución a la hora de prescribir este medicamento a pacientes con historial de úlcera péptica o gastritis. En pacientes con historial de isquemia miocárdica, debe administrarse con precaución. Se recomienda limitar el uso de otros medicamentos y alimentos que contengan cafeína cuando se esté en tratamiento con este medicamento. Precauciones o advertencias contiene acetaminofén/paracetamol. No usar con otros productos que contengan acetaminofén/paracetamol. El uso concomitante con otros productos que contienen acetaminofén puede conducir a una sobredosis. La sobredosis de acetaminofén puede causar falla hepática, la cual puede requerir trasplante de hígado o conducir a la muerte. Una enfermedad hepática existente incrementa el riesgo de daño hepático relacionado con el acetaminofén/paracetamol. Pacientes con diagnóstico previo de insuficiencia hepática o renal, deben consultar con el médico antes de tomar este medicamento. Casos de disfunción/falla hepática han sido reportados en pacientes con deficiencia en los niveles de glutatión, como aquellos con desnutrición severa, anoréxicos, con bajo índice de masa corporal, grandes consumidores crónicos de alcohol o que tienen sepsis. En pacientes con estados de deficiencia de glutatión, el uso de acetaminofén/paracetamol puede incrementar el riesgo de acidosis metabólica. Este medicamento contiene lactosa. Los pacientes con problemas hereditarios raros de intolerancia a la galactosa, deficiencia de lactasa lapp o malabsorción de glucosa-galactosa no deben tomar este medicamento.
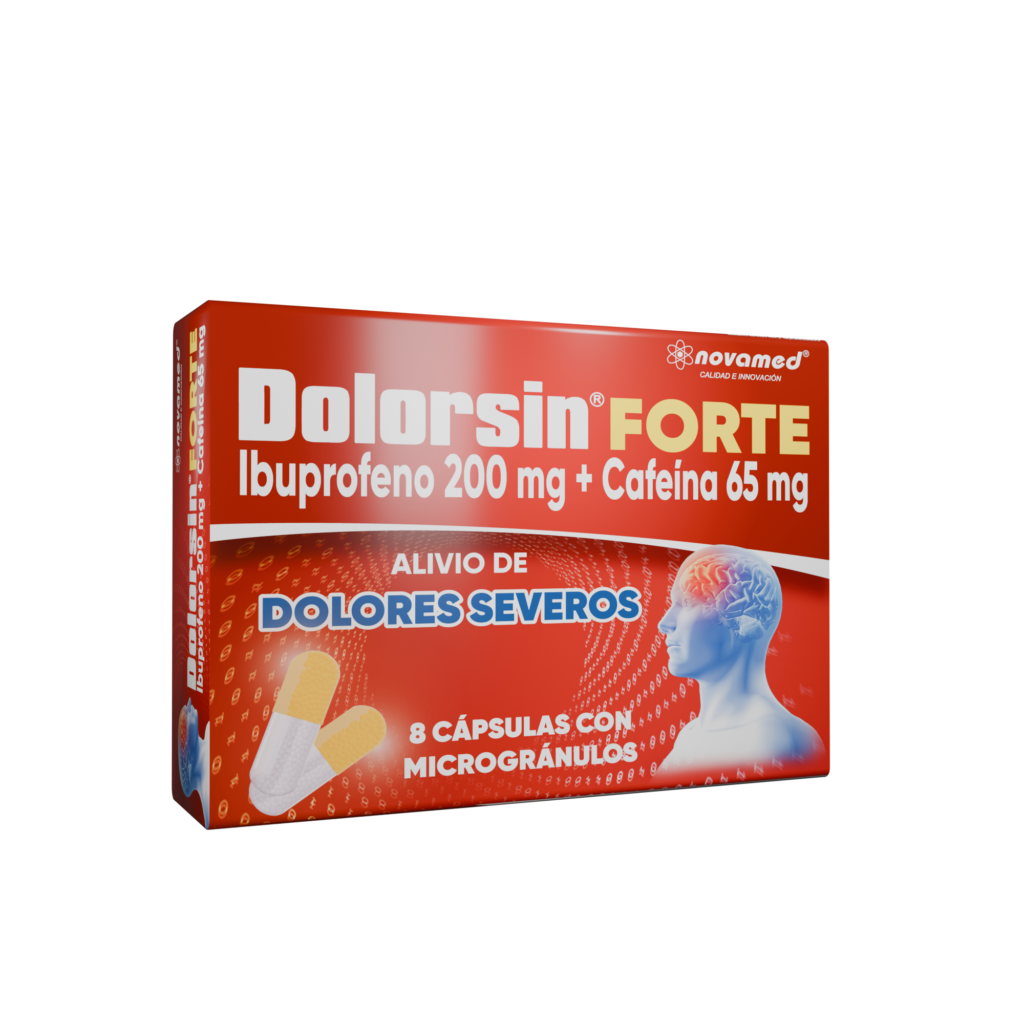
Dolorsin® Forte
REGISTRO SANITARIO:
INVIMA 2020M-0013927-R1
PRINCIPIO ACTIVO:
Ibuprofeno 200mg y Cafeína 65mg
UNIDAD DE VENTA:
Cápsula
Cápsulas
Analgésico anti-inflamatorio
no esteroide
(Ibuprofeno, cafeína)
COMPOSICIÓN: Cada cápsula contiene ibuprofeno 200 mg, cafeína 65 mg
INDICACIONES: Analgésico no narcótico y antipirético.
VÍA DE ADMINISTRACIÓN: Oral
FORMA FARMACÉUTICA: Cápsula
DOSIFICACIÓN: Adultos y niños mayores de 12 años: Tome una cápsula cada 6 a 8 horas. Si el dolor o la fiebre no responden a 1 cápsula, se pueden tomar 2 pero sin exceder de 6 en 24 horas. No se recomienda para menores de 12 años de edad.
CONTRAINDICACIONES: Reacciones alérgicas a la cafeína, al ibuprofeno o a los salicilatos u otros antiinflamatorios no esteroides (AINE) manifestadas por: Asma, broncoespasmo, rinitis aguda, pólipos nasales y edema agioneurótico o alergia a cualquier componente del producto. Personas que presenten desórdenes de la coagulación o que reciban anticoagulantes cumarínicos; enfermedad cardiovascular, falla renal; historia previa o actual de úlcera péptica o duodenal, sangrado gastrointestinal y enfermedad ácido péptica, insuficiencia hepática severa. No administrar durante el tercer trimestre del embarazo. ADVERTENCIAS Y PRECAUCIONES: Suspenda la administración y consulte a su médico, si nota una reacción alérgica que incluya enrojecimiento de la piel, rash o ampollas, si presenta vómito con sangre, sangre en las heces o heces negras. Consulte a su médico antes de administrarlo si usted tiene: Una enfermedad del corazón, hipertensión, una enfermedad renal, si está tomando otro antiinflamatorio no esteroide (AINE) u otro medicamento, si está embarazada o lactando, si usted está consumiendo ácido acetilsalicílico para la prevención de un infarto de miocardio (cardioprotector) o un accidente cerebro-vascular (ACV), debido a que el ibuprofeno puede disminuir el beneficio del ácido acetilsalicílico. La administración concomitante con ácido acetilsalicílico aumenta el riesgo de úlcera gastrointestinal y las complicaciones relacionadas. Se recomienda empezar el tratamiento con la dosis efectiva más baja. La administración continua a largo plazo puede incrementar el riesgo de eventos cardiovasculares y cerebrovasculares. Los efectos secundarios pueden ser minimizados con el uso de dosis bajas por periodos cortos de tiempo. Administrar con precaución en mayores de 60 años, pacientes con insuficiencia hepática moderada, insuficiencia renal grave (depuración de creatinina <30 mL/min). No se recomienda para menores de 12 años. Manténgase fuera del alcance de los niños. No exceda la dosis recomendada. Limite la administración de medicamentos, alimentos o bebidas que contengan cafeína. A menos que sea prescrito por un profesional del cuidado de la salud, detenga la administración y consulte si el dolor empeora o persiste por más de 10 días, o si la fiebre empeora o persiste por más de tres días. En caso de sobredosificación accidental, descontinuar la administración y consultar para asistencia médica inmediata.
INTERACCIONES: IBUPROFENO: El ibuprofeno debe administrarse con precaución en pacientes que estén en tratamiento con alguno de los fármacos que se citan a continuación ya que, en algunos pacientes, se han notificado interacciones:
Antihipertensivos: Reducción del efecto hipotensor. Diuréticos: Disminución del efecto diurético. Los diuréticos pueden aumentar el riesgo de nefrotoxicidad por antiinflamatorios no esteroideos. Glucósidos cardiacos: Los antiinflamatorios no esteroideos pueden exacerbar la insuficiencia cardiaca, reducir la tasa de filtración glomerular y aumentar los niveles de los glucósidos cardiacos. Litio: Disminución de la eliminación de litio. Metotrexato: Disminución de la eliminación de metotrexato. Ciclosporina: Aumenta el riesgo de nefrotoxicidad con los antiinflamatorios no esteroideos. Mifepristona: Los antiinflamatorios no esteroideos no deben administrarse en los 8-12 días posteriores a la administración de la mifepristona ya que éstos pueden reducir los efectos de la misma. Otros analgésicos: Evitar el uso concomitante con otros antiinflamatorios no esteroideos. Corticosteroides: Aumento del riesgo de sangrado digestivo. Anticoagulantes: Aumento del efecto anticoagulante. Quinolonas: Datos derivados de la experimentación animal indican que los antiinflamatorios no esteroideos asociados a las quinolonas pueden aumentar el riesgo de convulsiones. CAFEÍNA: Los efectos estimulantes de la cafeína pueden ser aditivos con los de otras sustancias que estimulan el sistema nervioso central. La cafeína debe ser evitada o utilizada con precaución si el paciente se encuentra medicado con metilfenidato, modafinilo, nicotina, pemolina, seudoefedrina, fenilpropanolamina o beta bloqueantes u otros simpaticomiméticos. La combinación de cafeína con estos fármacos puede producir nerviosismo, irritabilidad, insomnio y arritmias cardíacas. La administración concomitante de cafeína y fenilpropanolamina ha producido ocasionalmente accidentes cerebrovasculares, por lo que la administración conjunta de ambos fármacos debe ser evitada. Se ha descrito el desarrollo de síntomas psiquiátricos en pacientes tratados con clozapina después de la administración de cafeína. Las quinolonas antibacterianas (norfloxacina, ciprofloxacina, enoxacina, etc) reducen la eliminación de la cafeína, lo que puede originar un aumento exagerado de los efectos farmacológicos de esta sustancia. La cafeína reduce las concentraciones séricas de litio por lo que pacientes bajo tratamiento con este fármaco deberán ser advertidos de una posible reducción de los efectos antidepresivos del mismo. La cafeína interacciona igualmente con los inhibidores de la monoaminooxidasa (IMAOs) potenciando los efectos simpaticomiméticos de estos que pueden traducirse en arritmias cardíacas o grave hipertensión. El consumo de café esta contraindicado durante el tratamiento con IMAOs (p.ej. furazolidona, procarbazina, y selegilina) y durante 2 semanas después de la discontinuación de estos fármacos. Los pacientes tratados con benzodiazepinas para evitar el insomnio deben evitar el consumo de bebidas a base de cafeína poco antes de acostarse debido a que estas podrían antagonizar los efectos inductores del sueño de las primeras. Tampoco se deben administrar barbitúricos concomitantemente con cafeína, en primer lugar por ser los primeros inductores del metabolismo hepático con la consiguiente reducción de los niveles plasmáticos de la cafeína y en segundo lugar por poder anular la cafeína los efectos hipnóticos de los barbitúricos. Otros inductores del metabolismo hepático (fenitoína, rifampicina, etc) pueden igualmente reducir la eficacia de la cafeína. No se recomienda la administración concomitante de teofilina y de cafeína debido a la acumulación de efectos farmacológicos que podría conducir a una excesiva estimulación del sistema nervioso central con desarrollo de nerviosismo, irritabilidad, temblores, etc. En los neonatos, el uso conjunto de teofilina y cafeína puede resultar en el desarrollo de toxicidad
EFECTOS ADVERSOS: IBUPROFENO: Gastrointestinales: Son las reacciones adversas que se presentan con más frecuencia. Con la administración de ibuprofeno se ha notificado la aparición de náuseas, vómitos, diarrea, dispepsia, dolor abdominal, melenas, hematemesis, estomatitis ulcerosa y hemorragia digestiva. Se han observado con menor frecuencia gastritis, úlcera duodenal, úlcera gástrica y perforación. Los datos epidemiológicos indican que, de los siete antiinflamatorios no esteroideos más usados, el ibuprofeno posee el menor riesgo de toxicidad digestiva alta. Hipersensibilidad: Se han notificado reacciones de hipersensibilidad con ibuprofeno. Pueden consistir en (a) reacción alérgica inespecífica y anafilaxia, (b) reactividad del tracto respiratorio comprendiendo asma, agravación del asma, broncospasmo o disnea, o (c) alteraciones cutáneas variadas, incluyendo rash de varios tipos, prurito, urticaria, púrpura, angioedema y, menos frecuentemente, dermatosis bullosas (incluyendo necrólisis epidérmica y eritema multiforme). Cardiovasculares: Se ha notificado la aparición de edema asociada al tratamiento con ibuprofeno. Otras reacciones adversas que se han notificado con menor frecuencia y cuya relación no ha sido necesariamente establecida son: Renales: Varias formas de nefrotoxicidad, incluyendo nefritis intersticial, síndrome nefrótico e insuficiencia renal. Hepáticas: Alteración de la función hepática, hepatitis e ictericia. Neurológicas y de los órganos de los sentidos: Alteraciones visuales, neuritis óptica, cefalea, parestesias, depresión, confusión, alucinaciones, tinnitus, vértigo, mareo, fatiga y somnolencia. Se han descrito casos aislados de meningitis aséptica reversible al cesar el tratamiento. Su aparición es más probable en pacientes con lupus eritematoso y otras enfermedades del colágeno. Hematológicas: Trombocitopenia, neutropenia, agranulocitosis, anemia aplásica y hemolítica. Dermatológicas: Fotosensibilidad. CAFEÍNA: Las reacciones adversas de la cafeína son, en general, consecuencia de sus efectos farmacológicos. Cuando se administra en dosis terapéuticas, la cafeína puede ocasionar temblores, taquicardia sinusal y una mayor concentración mental. Otras reacciones adversas incluyen diarrea, excitación, irritabilidad, insomnio, tics musculares y palpitaciones Se han comunicado casos de hipoglucemia y de hiperglucemia después de dosis moderadas de cafeína. Con dosis más altas, se han descrito casos de náuseas y vómitos bastante intensos acompañados de ansiedad. Cuando las concentraciones plasmáticas de cafeína son > 50 mg/mL se producen síntomas de toxicidad, caracterizados por una intensificación de las anteriores reacciones adversas, acompañadas de arritmias cardíacas, convulsiones y delirios. La cafeína es un diurético moderado que puede producir poliuria. Se han comunicado casos de un aumento del aclaramiento de creatinina, hipercalciuria y un exceso de la eliminación urinaria de sodio.
PROPIEDADES FARMACODINÁMICAS: IBUPROFENO: Como todos los antiinflamatorios no esteroidicos de la familia de los ácidos aril-propionicos, el ibuprofen inhibe la acción de las enzimas COX-1 y COX-2. Los efectos anti-inflamatorios del ibuprofeno son el resultado de la inhibición periférica de la síntesis de prostaglandinas subsiguiente a la inhibición de la ciclooxigenasa. El ibuprofen inhibe la migración leucocitaria a las áreas inflamadas, impidiendo la liberación por los leucocitos de citoquinas y otras moléculas que actúan sobre los receptores nociceptivos. El ibuprofeno, como otros AINEs, no altera el umbral del dolor ni modifica los niveles de prostaglandinas cerebrales, concluyéndose que sus efectos son periféricos. La antipiresis es consecuencia de la vasodilatación periférica debido a una acción central sobre el centro regulador de la temperatura del hipotálamo. CAFEÍNA: Como potencializador para alivio de dolores severos, es un estimulante directo y moderado del sistema nervioso central y también estimula el corazón y el sistema cardiovascular. Le teofilina, estructuralmente parecida, comparte algunas de sus propiedades y se utiliza ampliamente en el tratamiento de enfermedades pulmonares. Tanto la cafeína como la que teofilina son estimulantes del sistema nervioso central siendo la segunda mucho más potente que la cafeína, particularmente a altas concentraciones. La cafeína también estimula el centro respiratorio medular y relaja el músculo bronquial liso. También estimula el músculo voluntario y la secreción gástrica de ácido, aumenta el flujo sanguíneo renal y tiene propiedades diuréticas moderadas. Mientras que las respuestas clínicas a la cafeína son bien conocidas, los mecanismos celulares son bastante inciertos. Se han propuesto varias teorías: A altas concentraciones la cafeína interfiere con la captación y el almacenamiento del calcio por el retículo sarcoplásmico del músculo estriado. Esta acción explicaría los efectos de la cafeína sobre el músculo cardíaco y esquelético pero no parece que las concentraciones necesarias se obtengan después de dosis clínicas. Actualmente se cree que las xantinas actúan como antagonistas de lo receptores de adenosina. La inhibición de las fosfodiesterasas y la acumulación subsiguiente de nucleótidos cíclicos tampoco parece que sea posible a las concentraciones clínicas. La adenosina actúa como un autacoide, y dado que prácticamente todas las células contienen receptores para la adenosina, los efectos clínicos son muy complejos. Inhibe la liberación de neurotransmisores y sus efectos pre- y post-sinápticos. El antagonismo de lo receptores de adenosina por la cafeína podría de esta manera promover la liberación de neurotransmisores, explicando los efectos estimulantes de la cafeína. Recientemente se ha descrito un síndrome asociado a la a la abstención a la cafeína. Es posible que las manifestaciones de la abstención a la cafeína sean secundarias a la depresión de las catecolaminas o de un neurotransmisor
CONDICIÓN DE VENTA: Venta libre
PRESENTACIÓN: Caja por x 32 cápsulas. Reg. San. INVIMA 2020M-0013927-R1.

Naproflash® Forte
REGISTRO SANITARIO:
. INVIMA 2015M- 0015805
PRINCIPIO ACTIVO:
Naproxeno 500mg y Cafeína 65mg
UNIDAD DE VENTA:
Tabletas
Analgésico, antipirético
(Naproxeno; Naproxeno
y cafeína)
COMPOSICIÓN: NAPROFLASH® FORTE, Cada tableta recubierta contiene naproxeno 500,00 mg, cafeína anhidra 65,00 mg, excipientes c.s.
INDICACIONES: NAPROFLASH: Analgésico, antipirético.
VÍA DE ADMINISTRACIÓN: Oral
FORMA FARMACÉUTICA: Naproflash® forte: Tableta
DOSIFICACIÓN: Adultos y niños mayores de 12 años: Tomar 1 o 2 cápsulas al inicio y continuar 1 cápsula cada 12 horas, sin exceder la dosis máxima en el día (24 horas) de 4 cápsulas. No se recomienda su uso por más de 3 días para el manejo de la fiebre, ni por más de 7 días para el manejo del dolor, excepto por prescripción médica. Sin los síntomas persisten o empeoran, consulte al médico. En caso de sobredosificación consultar al médico o al servicio de urgencias más cercano.
CONTRAINDICACIONES: (NAPROFLASH FORTE): Hipersensibilidad al principio activo o a sus excipientes. Broncoespasmo, rinitis aguda, pólipos nasales y edema angioneurótico. Reacciones alérgicas a ácido acetilsalicílico o aines. Úlcera péptica, sangrado gastrointestinal y antecedente de enfermedad ácido péptica. Disfunción hepática severa. Enfermedad cardiovascular, insuficiencia cardiaca congestiva, enfermedad coronaria, cirugía de derivación arterial coronaria (bypass), no administrar a mujeres en embarazo, en especial durante el tercer trimestre, en trabajo de parto, lactancia y niños menores de 12 años. PRECAUCIONES Y ADVERTENCIAS: advertencias: insuficiencia renal grave. (depuración de creatinina <30 ml/min). Insuficiencia hepática moderada. Se recomienda iniciar tratamiento con las dosis más bajas. El uso concomitante con el ácido acetil salicílico (asa) incrementa el riesgo de úlcera gastrointestinal y sus complicaciones. No tomar este medicamento si se han ingerido bebidas alcohólicas. Adminístrese con precaución a pacientes que reciben anticoagulantes cumarínicos e hipertensos con diuréticos, los cuales requieran estricto control médico. No tomar por más de 3 días para la fiebre y por más de 7 días para el dolor excepto por prescripción médica, si el dolor y la fiebre no ceden o empeoran se debe consultar al médico. Precauciones: teniendo en cuenta que la cafeína es sugestiva de promover ulceración gástrica, debe ser usada cautelosamente en pacientes con historia de ulcera péptica, además, por estar asociado con efectos arritmogénicos, es recomendado suspender este medicamento en personas con antecedente de síntomas de arritmias cardiacas, palpitaciones y en los primeros días y semanas posterior a un infarto de miocardio. Pacientes con alteración de la coagulación o que reciban fármacos que la alteren como los anticoagulantes (derivados heparinoides o dicumarol) deben observarse cuidadosamente sí reciben naproxeno.
INTERACCIONES: Naproxeno: Puede reducir el efecto antihipertensivo del propranolol y otros bloqueadores beta. Se recomienda precaución si naproxeno es administrado de manera concomitante con metotrexato. Los efectos estimulantes de la cafeína pueden ser aditivos con los de otras sustancias que estimulan el sistema nervioso central. Cafeína: Debe ser evitada o utilizada con precaución si el paciente se encuentra medicado con metilfenidato, modafinilo, nicotina, pemolina, pseudoefedrina, fenilpropanolamina, beta-bloqueantes u otros simpaticomiméticos.
EFECTOS SECUNDARIOS: Los pacientes con valores iniciales de hemoglobina de 10 gramos o menores, que van a recibir terapia prolongada, deben someterse a determinaciones periódicas de los valores de hemoglobina. Las actividades antipirética y antiinflamatoria de naproxeno pueden disminuir la fiebre e inflamación, disminuyendo así su utilidad como signos diagnósticos para detectar complicaciones de otros cuadros. Cafeína puede ocasionar temblores, taquicardia sinusal y una mayor concentración mental. Otras reacciones adversas incluyen diarrea, excitación, irritabilidad, insomnio, tics musculares y palpitaciones. Se han comunicado casos de hipoglucemia y de hiperglucemia después de dosis moderadas de cafeína. Con dosis más altas, se han descrito casos de náuseas y vómitos bastante intensos acompañados de ansiedad. PROPIEDADES FARMACODINÁMICAS: El naproxeno es un AINE derivado del ácido propiónico; es un inhibidor de la síntesis de prostaglandinas. Cafeína es coadyuvante en la disminución del dolor debido a que incrementa el efecto analgésico de los AINE, reduciendo efectos secundarios; la potenciación del efecto analgésico se debe a un cambio en la relación concentración del compartimiento-efecto del AINE, el cual está a una concentración efectiva 50 (EC50) y que incluye acciones a niveles central y periférico.
CONDICIÓN DE VENTA: NAPROFLASH® FORTE: Venta con fórmula médica.
PRESENTACIÓN:
NAPROFLASH® FORTE: Caja por 80 tabletas en blíster. Reg. San. INVIMA 2015M- 0015805). Es un medicamento, no exceder su consumo. Leer indicaciones y contraindicaciones en la etiqueta. Si los síntomas persisten consulte a su médico.

Vicitra®
REGISTRO SANITARIO:
INVIMA 2015M-0003721-R1
PRINCIPIO ACTIVO:
Vitamina C
UNIDAD DE VENTA:
Cápsulas
(Vitamina C
Ácido Ascórbico)
COMPOSICIÓN: Cada cápsula contiene Vitamina C 500 mg.
INDICACIONES: Deficiencia de vitamina C.
VÍA DE ADMINISTRACIÓN: Oral
FORMA FARMACÉUTICA: Capsulas
DOSIFICACIÓN: Niños de 2 a 6 años 1 cápsula al día, o según criterio médico. Niños de 6 a 12 años 1 a 2 cápsulas al día. Mayores de 12 años y adultos 2 a 4 cápsulas al día, máximo 4 cápsulas al día.
CONTRAINDICACIONES: Administrar con precaución en pacientes con oxaluria.
INTERACCIONES: El uso simultáneo de barbitúricos o primidona puede aumentar la excreción de ácido ascórbico en la orina. La acidificación de la orina que produce el uso de grandes dosis de ácido ascórbico puede acelerar la excreción renal de mexiletina. La prescripción conjunta con salicilatos aumenta la excreción urinaria de ácido ascórbico. El uso de ciertos medicamentos ocasiona depleción sérica de vitamina C (estrógenos, nicotina, ácido acetilsalicílico, alcohol, hierro, tetraciclinas). Se puede incrementar el riesgo de toxicidad por aluminio cuando se utiliza concomitantemente con los antiácidos, esto no es recomendado especialmente en pacientes con insuficiencia renal, si esto ocurre y no puede evitarse debe monitorearse el paciente para detectar posibles síntomas y signos de toxicidad aguda del aluminio (encefalopatía, ataques o coma) y proceder a retirar o ajustar su dosis. Ácido acetilsalicílico: Cuando se administran las drogas simultáneamente, ocurre un aumento en la excreción urinaria de ácido ascórbico y disminución de excreción de ácido acetilsalicílico. Se ha encontrado que ASA reduce la absorción del ácido ascórbico en cerca de un tercio. Dicumarol: Un solo caso aislado donde se reduce el tiempo de protombina luego de la ingesta de ácido ascórbico. Warfarina: Se ha reportado varios casos en los que el ácido ascórbico parece reducir el efecto de la warfarina. Estos reportes no fueron confirmados en pruebas subsiguientes. Etinilestradiol: Se ha reportado que el ácido ascórbico en dosis oral de 1g aumenta la biodisponibilidad del etinilestradiol en las preparaciones contraceptivas orales. Este efecto puede ser importante si se discontinua el suplemento de ácido ascórbico, ya que la caída en la absorción de la hormona puede llevar sangrado ruptura o hasta falla contraceptiva. Sin embargo, no existen estudios en este efecto del etinilestradiol cuando la vitamina C ha sido administrada de forma intravenosa o como inyección intramuscular. Hierro (oral): El ácido ascórbico oral puede incrementar la absorción de hierro. Sin embargo, los mecanismos de retroalimentación generalmente controlan su absorción excesiva. Deferoxamina: El ácido ascórbico puede aumentar la excreción de hierro cuando se da en forma concomitante con la deferoxamina, sin embargo, han ocurrido casos de cardiomiopatía y falla cardiaca congestiva en pacientes con tratamiento concomitante. Puede ser que el ácido ascórbico movilice el hierro del bazo y de otros tejidos reticuloendoteliales resultando en el aumento de disposición de hierro en los órganos viscerales. En general, se recomienda que la dosis de ácido ascórbico sea administrada una o dos horas luego de que haya empezado la infusión de deferoxamina. Isoprenalina: El efecto cronotrópico de la isoprenalina disminuye cuando se administra en forma simultánea con el ácido ascórbico. Alcohol: El alcohol reduce los niveles de ácido ascórbico. Disulfiram: El uso crónico de altas dosis de ácido ascórbico puede interferir con la interacción de disulfiram–alcohol cuando se usan de forma simultánea. Mexiletina: Altas dosis de ácido ascórbico pueden acelerar la excreción renal de la mexiletina cuando las drogas se administran en forma simultánea. Barbitúricos o primidona: La excreción urinaria de ácido ascórbico puede aumentar cuando se administra junto con barbitúricos o primidona. Flufenazina y otras fenotiazinas: Se ha reportado que el ácido ascórbico disminuye el efecto terapéutico de las fenotiazinas. La concentración de flufenazina también puede ser reducida. Anfetaminas y antidepresivos tricíclicos: El ácido ascórbico disminuye la reabsorción tubular renal de las anfetaminas y los antidepresivos tricíclicos.
EFECTOS SECUNDARIOS: En dosis altas puede producir precipitación de cálculos de oxalato en el tracto urinario y presentar dolor en la zona renal. Diarrea, cefaleas, náuseas, vómitos y gastralgias son síntomas raros que pueden aparecer por dosis elevadas. En algunos casos aislados después de la administración prolongada de 2 a 3 g por día, se puede producir escorbuto al retirar la medicación.
PROPIEDADES FARMACODINÁMICAS: El ácido ascórbico es esencial en múltiples procesos del metabolismo y la respiración celular y en los sistemas de óxido-reducción de la membrana celular. Actúa en la síntesis de colágeno, hueso, cartílago, piel y dientes. Al estimular los sistemas de defensa natural contra enfermedades infecciosas mejora la resistencia natural a las enfermedades.
CONDICIÓN DE VENTA: Venta Libre
PRESENTACIÓN: Caja por 30 unidades. Cada cápsula contiene 500 mg de vitamina C. Reg. San. INVIMA 2015M-0003721-R1.

Vicitra® Z
REGISTRO SANITARIO:
INVIMA SD2021-0004588
PRINCIPIO ACTIVO:
Vitamina C 500mg y Zinc 5mg
UNIDAD DE VENTA:
Cápsulas



CDZ Vit®
La Vitamina C contribuye al funcionamiento normal del sistema inmune. CDZ Vit® contiene 1000 mg de Vitamina C.
La Vitamina C contribuye al funcionamiento normal del sistema inmune durante y después del ejercicio físico intenso. CDZ Vit® contiene 1000 mg de Vitamina C.
La Vitamina D contribuye al funcionamiento normal del sistema inmune.
CDZ Vit® contiene 1000 UI de Vitamina D. El Zinc contribuye al funcionamiento normal del sistema inmune.
CDZ Vit® contiene 40 mg de Zinc. Una ingesta adecuada de Zinc ayuda a fortalecer las defensas del organismo.
CDZ Vit® Contiene 40 mg de Zinc.

Neuromed®
REGISTRO SANITARIO:
INVIMA SD2021-0004630
PRINCIPIO ACTIVO:
Lecitina (no modificada genéticamente) 760 mg, fosfatidilcolina 280 mg, fósforo 12 mg
UNIDAD DE VENTA:
Cápsulas